
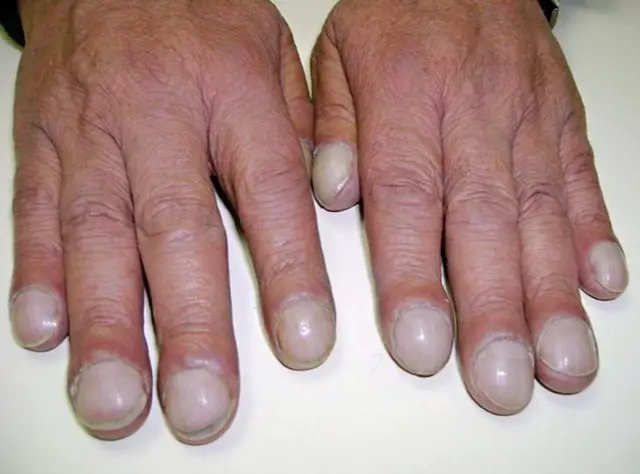
Le dita ippocratiche sono uno dei segni di fibrosi cistica
La fibrosi cistica è una rara malattia genetica autosomica recessiva causata da una mutazione di un gene sito sul cromosoma 7, il quale codifica per una proteina che funziona come canale per il cloro, detta CFTR (Cystic Fibrosis Transmembrane conductance Regulator). La fibrosi cistica, un tempo mortale nel primo anno di nascita, è oggi una malattia che può essere curata, tuttavia rimane una malattia fortemente debilitante, che abbassa la qualità della vita del paziente e diminuisce la sua aspettativa di vita.
Epidemiologia
Questa malattia costituisce il più comune disordine genetico ereditario letale negli Stati Uniti, in quanto colpisce fino ad 1 bambino su 2000 (1 su 4000 secondo altri autori). La fibrosi cistica si riscontra nei maschi e nelle femmine allo stesso modo, ma i maschi tendono ad avere una speranza di vita più lunga rispetto alle femmine, forse perché l’ormone femminile estrogeno è capace di peggiorare le condizioni della malattia. La fibrosi cistica viene tipicamente diagnosticata in età infantile. La fibrosi cistica compare principalmente in individui bianchi di origine europea: tra gli asiatici e gli africani la malattia è molto meno diffusa.
Storia
La fibrosi cistica è stata per la prima volta segnalata nel 1936, quando Fanconi e coll. descrissero due bambini con”fibrosi cistica del pancreas e bronchiectasie“. Ad ogni modo si pensa che la fibrosi cistica sia stata probabilmente altrettanto frequente e letale nei secoli precedenti la pubblicazione dell’articolo fondamentale da parte di Fanconi. Nel XVIII e XIX secolo, infatti, nella letteratura europea sono comparse numerose segnalazioni di bambini affetti da anomalie che, lette con le conoscenze attuali, sono fortemente suggestive di fibrosi cistica. Gran parte di queste segnalazioni evidenziava la correlazione tra un sapore salato della cute di questi bambini quando venivano baciati e la probabilità di decesso del bambino in età molto precoce.
Organi coinvolti
La fibrosi cistica determina il malfunzionamento di numerose strutture anatomiche. Gli organi coinvolti nella fibrosi cistica e le malattie relative a tali organi, sono principalmente:
- Polmoni
- Bronchiectasie
- Bronchiti
- Polmoniti
- Atelettasie
- Tappi di muco
- Insufficienza respiratoria
- Pancreas
- Insufficienza pancreatica esocrina
- Pancreatiti ricorrenti
- Diabete mellito
- Ghiandole sudoripare
- Aumentata concentrazione di elettroliti nel sudore
- Vie aeree superiori
- Sinusiti ricorrenti
- Polipi nasali
- Intestino
- Tappi di meconio
- Ileo da meconio
- Intussuscezione
- Fegato
- Cirrosi
- Ittero neonatale
- Colecisti
- Colelitiasi
- Ghiandole salivari
- Alterata concentrazione di elettroliti nelle secrezioni
- Sistema riproduttivo
- Ostruzione del dotto deferente
- Ridotta fertilità femminile.
I problemi più importanti associati a tale malattia sono le bronchiectasie (anomala dilatazione dei bronchi), l’insufficienza pancreatica esocrina e la presenza di una elevata concentrazione elettrolitica nel sudore. L’interessamento patologico del polmone crea i maggiori problemi nei pazienti con fibrosi cistica e costituisce la principale causa di decesso.
Trasmissione
Le caratteristiche di ereditarietà sono di tipo autosomico recessivo. Una malattia è detta a trasmissione autosomica recessiva quando l’allele alterato deve essere presente in coppia (omozigosi), cioè sono necessarie due copie dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso. Non basta un solo genitore portatore sano o malato, bensì entrambi i genitori devono essere portatori sani o malati. Il fenotipo quindi si esprime quando nel genotipo dell’individuo sono presenti entrambi gli alleli responsabili, fatto che spiega l’alta probabilità di sviluppare malattie genetiche in caso di incesto. Quindi:
- un individuo che possegga entrambi gli alleli alterati: è portatore ed è malato;
- un individuo che possegga solo un allele alterato: è portatore ma è sano;
- un individuo che non possegga nessun allele alterato: NON è portatore ed è sano.
Essere portatore sano vuol dire quindi NON avere la patologia ma possedere nel proprio genotipo un allele mutato, che può essere trasmesso alle generazioni successive.
Dalla combinazione delle possibili condizioni di genitori sani, malati e portatori sani, deriva la distribuzione probabilità che la malattia sia trasmessa ai figli:
- genitori malato-malato: la probabilità che il figlio/a nasca malato è del 100%;
- genitori sano-malato: la probabilità che il figlio/a nasca portatore sano è del 100%;
- genitori malato-portatore sano: la probabilità che il figlio/a nasca malato è del 50% e del 50% che nasca portatore sano;
- genitori sano-portatore sano: la probabilità che il figlio/a nasca sano è del 50% e del 50% che nasca portatore sano;
- genitori portatore-portatore: la probabilità che il figlio/a nasca portatore sano è del 50% mentre è del 25% che nasca sano o malato.
Se nessuno dei genitori ha un allele mutato, non c’è ovviamente alcuna trasmissione autosomica recessiva ed i figli saranno tutti sani e NON portatori dell’allele mutato.
Nell’immagine che segue, è raffigurata la tipica situazione in cui entrambi i genitori sono sani ma portatori dell’allele mutato:
- un figlio su quattro avrà entrambi gli alleli alterati e sarà malato ed ovviamente portatore;
- due figli su quattro avranno un allele normale ed uno alterato e saranno sani ma anche portatori;
- un figlio su quattro avrà entrambi gli alleli normali e sarà sano e NON portatore.

Cause e fattori di rischio
La causa della fibrosi cistica è una alterazione genetica che riguarda il gene CFTR, che si trova al locus q31.2 del cromosoma 7. Esso può essere alterato (mutato) in almeno 20 differenti modi e molte di queste mutazioni sono causa di fibrosi cistica. Si pensa che da 1:16 a 1:25 americani di razza bianca siano portatori di un gene CFTR alterato. La frequenza di tale gene nella popolazione orientale e nei neri è, invece, molto inferiore a quella evidenziata nei bianchi per cui, conseguentemente, la malattia si verifica nei soggetti non di razza bianca con frequenza molto minore. L’anomalia più comune del gene è la delezione di 3 paia di basi nella catena del DNA, la quale porta alla perdita di un aminoacido dalla proteina codificata dal gene. Tale mutazione è nota come delta F;08 e rende conto del 70% delle anomalie genetiche responsabili della fibrosi cistica. La severità della fibrosi cistica è in parte legata alla forma genetica della malattia ereditata dal paziente.
Il gene della fibrosi cistica contiene le informazioni per l’elaborazione di una grande proteina che regola il flusso di ioni (sali) attraverso le ghiandole che secernono i liquidi (ghiandole esocrine): in conseguenza di ciò i pazienti con fibrosi cistica non sono in grado di regolare la composizione dei sali nelle proprie secrezioni come i soggetti
normali. Questa errata regolazione della secrezione è responsabile della maggior parte, se non di tutti, i problemi manifestati dai soggetti affetti da fibrosi cistica.
Il fattore di rischio più importante per la fibrosi cistica è ovviamente l’avere un genitore malato di fibrosi cistica o avere uno o entrambi i genitori portatori sani di fibrosi cistica.
Anatomia patologica e fisiopatologia
La fibrosi cistica viene considerata come una endocrinopatia generalizzata. In un certo senso, virtualmente, tutte le ghiandole esocrine del corpo sono colpite dalla malattia. La fibrosi cistica si manifesta con una classica triade di anomalie esocrine. Queste anomalie sono rappresentate da insufficienza pancreatica, infezioni polmonari croniche ricorrenti ed elevata concentrazione degli elettroliti nel sudore. Nei pazienti molto giovani affetti da fibrosi cistica il pancreas appare normale. Con il progredire della malattia il pancreas diviene più piccolo e fibrotico. L’esame microscopico del pancreas rivela l’ostruzione dei dotti e dei duttuli pancreatici. Successivamente si verifica la dilatazione del lume ghiandolare e, alla fine, la sostituzione del tessuto ghiandolare esocrino con tessuto connettivo fibrotico.
La pneumopatia è responsabile della maggiore morbidità e mortalità dei pazienti affetti da fibrosi cistica. I tre problemi di natura polmonare più frequentemente riscontrati nei pazienti con fibrosi cistica sono le infezioni polmonari ricorrenti, le bronchiectasie e l’iperattività bronchiale. Questi problemi possono essere di lieve entità nei bambini,ma
le loro incidenze e gravità aumentano con il progredire del processo patologico. Nelle fasi precoci della malattia i polmoni appaiono spesso normali. Nel momento in cui il paziente comincia a svilupparsi i polmoni mostrano un progressivo incremento delle dimensioni e del numero delle goblet cells (ghiandole mucose), oltre all’infiammazione del tessuto peribronchiale. La mucosa delle vie aeree si modifica dal normale epitelio in quello squamoso stratificato, mediante un processo definito metaplasia squamosa. Nei pazienti che hanno manifestato sintomi polmonari per diversi anni, questi reperti sono più diffusi e gravi. Le modificazioni enfisematose sono di frequente riscontro, inoltre l’emorragia può essere osservata all’interno dei polmoni. I tappi mucosi si osservano nelle piccole vie aeree mentre le bronchiectasie costituiscono un reperto ubiquitario.
Durante le fasi terminali della malattia è frequente il riscontro dell’enfisema ostruttivo, sebbene la distruzione coinvolga raramente più del 10% del tessuto polmonare. I tappi mucosi sono diffusi e gli ascessi si riscontrano nella porzione distale dei polmoni. I linfonodi ilari sono ingrossati.
Per approfondire:
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cureFibrosi cistica: sintomi, segni, diagnosi, esami
- Fibrosi cistica: terapia farmacologica e chirurgica, prognosi, mortalità
Leggi anche
- Differenza tra malattia autosomica dominante e recessiva con esempi
- Le malattie genetiche più diffuse al mondo
- Insufficienza respiratoria acuta e cronica: cause e conseguenze
- Insufficienza polmonare lieve, severa, acuta: sintomi e cura
- Differenza tra insufficienza respiratoria di tipo 1 e 2
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- I migliori saturimetri professionali per uso ospedaliero e casalingo
- Emogasanalisi arterioso: procedura, interpretazione, è dolorosa?
- Ipossiemia: significato, valori, sintomi, conseguenze, rischi, cure
- Ipercapnia: valori, terapia, conseguenze e trattamento
- Ipossia: valori, conseguenze, sintomi, cure
- Anossia: definizione, cause, sintomi, sinonimo, cure
- Ipocapnia: significato, cause, valori, alcalosi respiratoria
- Differenza tra ipossiemia e ipercapnia
- Differenza tra ipossiemia, ipossia, anossiemia ed anossia
- Alterazioni dell’equilibrio acido-base: acidosi ed alcalosi respiratorie e metabolica
- Differenza tra acidosi ed alcalosi, metabolica e respiratoria
- Sindrome da distress respiratorio (ARDS): definizione e linee guida
- Edema polmonare acuto, cardiogeno, cause, sintomi e terapie
- Radiografia del torace: come si fa, indicazioni, bisogna spogliarsi, costo
- Broncopneumopatia cronica ostruttiva (BPCO): sintomi, diagnosi e cura
- Broncoscopia polmonare con biopsia: a cosa serve, fa male, è pericolosa?
- Tumore al polmone: aspettativa di vita, sopravvivenza, fase terminale
- Tumore pleurico e mesotelioma: sopravvivenza e aspettativa di vita
- Cavo, liquido e versamento pleurico: fisiologia e patologia
- Differenza tra PM10 e PM2,5 e rispettivi effetti sulla salute
- Da oggi l’inquinamento dell’aria è ufficialmente un cancerogeno
- Polvere toracica: le case dei fumatori sono inquinate come Pechino
- Consigli per la prevenzione delle sindromi asmatiche
- Attività fisica e asma bronchiale: sport consigliati e sconsigliati
- Asma bronchiale estrinseca, intrinseca, occupazionale, stabile: cause, sintomi, cure
- Asma bronchiale in bambini e adulti: cause, sintomi e cura
- Test di provocazione bronchiale con metacolina: esecuzione, preparazione, rischi
- Iper-reattività bronchiale: significato, sintomi, diagnosi e cure
- Asma occupazionale: cause, sintomi, diagnosi e terapie
- Malattie respiratorie occupazionali o professionali: effetti, elenco, tipi
- Visita allergologica: svolgimento, esami, preparazione, durata, costo
- Differenza tra allergia, pseudoallergia e intolleranza alimentare
- Consigli per la prevenzione delle allergie inalatorie da pollini
- Broncopolmonite: sintomi iniziali, contagio, prognosi, morte
- Polmonite ab ingestis: cause, tempi di guarigione, morte, sopravvivenza
- Tracheite virale, batterica, allergica: quanto dura e come si cura
- Laringite acuta e cronica: antibiotico, catarrale, cure, rimedi naturali
- Faringite virale e batterica: rimedi, quanto dura, è contagiosa?
- Bronchite: sintomi, durata, cura, rimedi naturali, è contagiosa?
- Differenza tra bronchite acuta e cronica
- Differenza tra bronchite e polmonite
- Mal di gola forte: rimedi naturali e farmaci per farlo passare
- Cordite in medicina: significato, da reflusso, sintomi, cura
- Laringoscopia diretta e indiretta: anestesia, costo, è dolorosa?
- Differenza tra laringoscopia diretta, indiretta, rigida, flessibile
- Intossicazione da monossido di carbonio: sintomi, danni permanenti, morte
- Atelettasia: significato, polmonare, cause, sintomi, cura, riabilitazione
- Laringospasmo: virale, da ansia, da stress, significato
- Broncospasmo in bambini e anziani: rimedi, quanto dura, paradosso
- Differenza tra laringospasmo e broncospasmo
- Morte per soffocamento: segni, sintomi, fasi e tempi
- Cosa si prova a morire annegati, dissanguati, decapitati…
- Differenze tra faringite, laringite e tracheite: vari tipi di mal di gola
- Laringite ipoglottica (pseudocroup): cause, sintomi, cosa fare
- Croup nel bambino: significato, cause, sintomi, terapia, mortalità
- Differenza tra croup e pseudocroup
- Bronchiolite in neonati e bambini: sintomi, cause, è pericolosa?
- Bronchiolite nei bimbi: mortalità, pericoli, complicazioni e durata
- Tampone faringeo: preparazione, digiuno, positivo o negativo
- Differenza tra tonsillite, faringite, laringite, tracheite, adenoidite
- Differenza tra mal di gola e tonsillite
- Differenza tra mal di gola virale e batterico
- Abbassamento della voce: cause, rimedi, quando andare dal medico
- Polipi, noduli e granulomi delle corde vocali: cause, sintomi e cure
- Dove si trovano le corde vocali: anatomia e funzioni in sintesi
- Sensazione di nodo alla gola in medicina: cause, sintomi, diagnosi, cure
- Perché viene la tosse e come faccio a farla passare?
- Perché si starnutisce? Cosa fare se si continua a starnutire?
- Differenza tra tosse secca, grassa, cronica e con catarro
- Perché ci viene la febbre e perché non dobbiamo aver paura di lei
- Differenza tra raffreddore e influenza: sintomi comuni e diversi
- Come e quando abbassare la febbre? Farmaci e rimedi casalinghi
- Tonsille gonfie, infiammate e con placche rimedi in adulti e bambini
- Tonsillectomia: cosa mangiare, convalescenza, dolore, in adulti e bambini
- Tonsille palatine, linguali, faringee e tubariche: funzioni e patologia
- Differenza tra tonsille e adenoidi
- Adenoidi: ingrossate, operazione, convalescenza, ricrescono?
- Adenoidite acuta e cronica in bambini ed adulti: cause, sintomi, cure
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Asfissia: sintomi, cure ed in quanto tempo si muore
- Respirazione artificiale bocca a bocca: quando farla e come farla
- Primo soccorso e BLS (Basic Life Support): cos’è e come si fa
- Cianosi in volto, mani o labbra: significato, cause, rischi, cure
- Differenza tra Mucosolvan e Fluimucil
- Spirometria diretta ed indiretta: come si esegue ed a cosa serve
- Sì al bagno dopo mangiato, ecco le vere 8 cause più frequenti di morte in acqua
- Croup nel bambino: significato, cause, sintomi, terapia, mortalità
- Differenza tra asfissia e soffocamento
- Differenza tra soffocamento e strangolamento
- Massaggio cardiaco: quando farlo e come farlo [LINEE GUIDA]
- Morte in culla (SIDS): prevenzione, cause, sintomi e percentuale dei casi
- Malattia da decompressione: terapia e fisiopatologia
- Embolia gassosa arteriosa da immersione: sintomi e cure
- Cosa succede al cibo nello stomaco dopo averlo ingerito?
- Apnea ostruttiva del sonno: cause, rischi, trattamenti e prevenzione
- Perché si russa e quali sono i rimedi per smettere di russare? I pericoli dell’apnea ostruttiva del sonno
- Crisi respiratoria acuta e rischio di morte: cosa fare?
- Pallore in viso: significato, sinonimo, cause, ansia, cosa mangiare
- Le 7 fasi della deglutizione (volontarie ed involontarie)
- Differenza tra disfagia di tipo ostruttivo e di tipo motorio
- Differenza tra disfagia ai liquidi e ai solidi
- Insufficienza respiratoria acuta e cronica: cause e conseguenze
- Intubazione: rischi, anestesia, rianimazione, dolore alla gola
- Tracheotomia possibilità di parlare, durata, conseguenze, quando si fa
- Tracheostomia: complicanze, parlare, percutanea, cibo e gestione
- Differenza tra tracheostomia percutanea e tradizionale (a cielo aperto)
- Differenza tra tracheotomia e tracheostomia
- Cricotiroidotomia: urgenza, complicanze, procedura ed indicazioni
- Differenza tra tracheotomia e cricotiroidotomia
- Come si misura la frequenza respiratoria?
- Frequenza respiratoria normale, alta, bassa, a riposo e sotto sforzo
- Differenze tra respiro normale e patologico
- Iperventilazione: significato, sintomi, alcalosi e conseguenze
- Sindrome da iperventilazione cronica: cause, sintomi, diagnosi, cure
- Dispnea ansiosa, notturna e cardiaca: sintomi, diagnosi e cura
- Differenza tra dispnea, apnea e tachipnea
- Enfisema polmonare: sintomi, tipi, cause, diagnosi e terapia
- I migliori elettrocardiografi ed Holter portatili
- I migliori sfigmomanometri e stetoscopi per misurare la pressione arteriosa
- I migliori glucometri di ultima generazione per misurare la glicemia
- I migliori misuratori di colesterolo LDL HDL e trigliceridi, affidabili e precisi
- Fenomeno di Raynaud: cause, sintomi e trattamento
- Cos’è l’edema, come e perché si forma?
- Pneumotorace spontaneo primario, secondario ed iperteso: cause, sintomi, terapie
- Dipendenza da nicotina: come smettere di fumare sigarette in 20 passi
- Stop alle sigarette: i migliori farmaci per smettere di fumare
- Champix compresse per smettere di fumare: foglio illustrativo, prezzo
- Bupropione per smettere di fumare: meccanismo d’azione, posologia
- Gomme e cerotti: i sostituti con nicotina per smettere di fumare
- Gomme, farmaci, cerotti… Qual è il metodo migliore per smettere di fumare?
- Smettere di fumare: meglio diminuire le sigarette o interrompere di colpo?
- Vareniclina per emettere di fumare: meccanismo d’azione, efficacia
- Champix: funziona davvero per smettere di fumare?
- Respiro patologico: le alterazioni del ritmo respiratorio normale
- Respiro di Biot ed apnee: caratteristiche e cause patologiche e non patologiche
- Respiro di Cheyne-Stokes: caratteristiche e cause patologiche e non patologiche
- Respiro di Falstaff: caratteristiche e cause
- Respiro di Kussmaul: caratteristiche e cause
- Idrotorace: cause, patologie, sintomi, diagnosi e cure
- Embolia polmonare: massiva, diagnosi, da tumore, terapia
- Polmoniti nosocomiali: cause, terapie e linee guida ATS
- Polmonite interstiziale, atipica, senza febbre: sintomi e cure in bimbi ed adulti
- Tumore al polmone operabile ed inoperabile: stadiazione
- Massaggio cardiaco: quante compressioni al minuto?
- Toracentesi: procedura, complicanze, rischi, è dolorosa?
- Parametri della spirometria: capacità, volumi, rapporti e flussi
- Empiema pleurico: guarigione, saccato, complicanze, RX, cura
- Differenza tra dispnea ed affanno
- Differenza tra polipnea e tachipnea
- Tipologie di respirazione nello yoga
- Apparato respiratorio: anatomia in sintesi, struttura e funzioni
- Trachea: anatomia e funzioni in sintesi
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Differenza tra inspirazione e espirazione: l’atto respiratorio
- Differenza tra ventilazione polmonare e alveolare: spazio morto anatomico e fisiologico
- I polmoni fanno male: i sintomi di una malattia polmonare
- Dove si trovano i polmoni ed a che servono?
- Esaminare il polmone con le prove di funzionalità respiratoria
- Ipertensione polmonare: lieve, severa, terapia, aspettativa di vita
- Si può vivere senza uno o entrambi i polmoni?
- Si può vivere senza respirare aria? Quanto può durare una apnea?
- Si può vivere di sola aria senza mangiare né bere? Per quanto tempo?
- Ossigenoterapia: uso, controindicazioni, domiciliare, con maschera
- Ventilazione meccanica o artificiale: tipi ed indicazioni
- Differenza tra ventilazione meccanica e ossigenoterapia
- Pallone AMBU: a cosa serve, componenti, scadenza, percentuale di ossigeno
- Pallone va e vieni: cos’è, come funziona, a che serve
- Differenza tra pallone AMBU e va e vieni: vantaggi e svantaggi
- Cannula nasale per ossigenoterapia: cos’è, come è fatta, quando si usa
- Sondino nasale per ossigenoterapia: cos’è, come è fatta, quando si usa
- Differenza tra NIV, CPAP e BIBAP
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
