 La sindrome di Cornelia de Lange (CDLS) è una sindrome malformativa riconosciuta per la prima volta nel 1933 da una pediatra che diede ad essa il nome, la cui incidenza è stimata pari ad 1 nuovo caso ogni 10-20.000 nati (circa 50 nuovi nati all’anno in Italia). Una sindrome malformativa è una malattia congenita (cioè presente sin dalla nascita) caratterizzata dalla possibile presenza contemporanea di malformazioni maggiori (alterazione della struttura di cuore, reni, palato, sistema nervoso centrale, arti, genitali esterni ecc.), di problemi di crescita in peso e altezza (in eccesso o in difetto), di anomalo sviluppo psicomotorio e intellettivo e di tratti somatici spesso molto particolari.
La sindrome di Cornelia de Lange (CDLS) è una sindrome malformativa riconosciuta per la prima volta nel 1933 da una pediatra che diede ad essa il nome, la cui incidenza è stimata pari ad 1 nuovo caso ogni 10-20.000 nati (circa 50 nuovi nati all’anno in Italia). Una sindrome malformativa è una malattia congenita (cioè presente sin dalla nascita) caratterizzata dalla possibile presenza contemporanea di malformazioni maggiori (alterazione della struttura di cuore, reni, palato, sistema nervoso centrale, arti, genitali esterni ecc.), di problemi di crescita in peso e altezza (in eccesso o in difetto), di anomalo sviluppo psicomotorio e intellettivo e di tratti somatici spesso molto particolari.
Segni e sintomi in generali
Le caratteristiche principali di un soggetto affetto sono costituite da basso peso alla nascita (inferiore a due chili e mezzo), scarsa crescita postnatale in peso e altezza, ridotte dimensioni della testa (microcefalia), eccessiva peluria sul corpo (avambraccio e regione lombare in particolare), mani e piedi piccoli o gravi alterazioni malformative a carico delle mani stesse. I pazienti si assomigliano molto tra di loro: le ciglia sono lunghe, il naso è piccolo con narici all’insù, le labbra sono sottili con angoli rivolti in basso. Le sopracciglia sono sottili, arcuate e unite frequentemente sulla linea mediana e danno l’impressione di essere state disegnate con la matita. L’acquisizione delle comuni tappe di sviluppo di ogni bambino (stare seduto, gattonare, camminare, dire le prime parole) è ritardata, così come ritardato in modo variabile è lo sviluppo intellettivo. Per quanto riguarda infine l’area della comunicazione verbale (linguaggio), si tratta della sfera più compromessa ed è ormai chiaro che le capacità di comprensione dei soggetti affetti superano notevolmente le loro possibilità di espressione verbale.
Leggi anche: Cos’è un cromosoma ed a che serve?
Crescita e alimentazione
Altezza, peso e circonferenza cranica durante l’infanzia di solito sono al di sotto del terzo percentile. Individui con la forma lieve della sindrome hanno un ritardo di crescita meno severo. Lo scatto di crescita puberale si ha in tempi normali (una media di 15 anni per i maschi e di 13 anni per le femmine), ma l’ampiezza dello scatto di crescita e inferiore a quella della popolazione generale. L’altezza media, da adulti, e di 155.78 cm per i maschi e di 131.13 per le femmine. Fin dalla nascita, la capacita di nutrirsi e scarsa, e non di rado i bambini affetti necessitano di alimentazione per via nasogastrica. In qualche e necessaria l’inserzione un tubo per gastrostomia. E comune il reflusso gastroesofageo, che contribuisce alle difficolta di alimentazione e alla scarsa crescita. La prognosi per una crescita normale e buona se il sottostante reflusso gastroesofageo e diagnosticato e trattato precocemente.
Sviluppo e comportamento
Numerosi studi e case report hanno documentato il QI dei soggetti affetti, e i risultati variavano da 30 a 60 per gli individui con la forma classica, fino a 102 in quelli con la forma lieve. Barney et al. Nel 1999, condussero uno studio dettagliato sul comportamento di 49 individui con i fenotipi classico e lieve. L’età dei pazienti dello studio variava della prima infanzia all’età adulta. Essi evidenziarono una grande varietà di sintomi, che includevano l’iperattività (40%), l’autolesionismo (44%), aggressioni quotidiane (49%) e disturbi del sonno (55%). I disturbi del comportamento correlavano strettamente con la presenza di una sindrome simil-autistica e con il grado di ritardo mentale. Questi autori descrissero come questi disturbi del comportamento continuassero anche in età adulta. Kline e colleghi misero a punto una valutazione dello sviluppo in 122 individui con sindrome di Cornelia de Lange, usando una varietà di metodi, e furono in grado di costruire tavole con le tappe di sviluppo, prendendo in considerazione la motricità grossolana, la motricità fine, le abilita sociali e lo sviluppo del linguaggio. Vi sono molte descrizioni, in letteratura, di assenza di linguaggio negli individui con la sindrome di Cornelia de Lange. All’altro estremo dello spettro, vi e la descrizione di una ragazza con la forma lieve della sindrome che a 14 anni era in grado di parlare con frasi articolate. In uno studio sugli indicatori prognostici per lo sviluppo del linguaggio, i fattori che predicevano una prognosi scarsa erano: un peso alla nascita al di sotto 2.270 kg, deficit uditivo da moderato a grave, malformazioni a carico degli arti superiori, scarse interazioni sociali e un grave ritardo motorio. Molti lavori hanno descritto un pianto simile ad un ringhio sommesso come caratteristico della sindrome in infanzia. Non e stato possibile valutare gli effetti della sordità combinata sensitiva e conduttiva, che si osserva nella sindrome, sul deficit di linguaggio, perché nessuno degli studi sulla sindrome ha incluso una valutazione dell’udito. Poiché adesso gli individui affetti dalla sindrome di Cornelia de Lange sono trattati con dispositivi di ausilio all’udito e con l’inserzione di tube di ventilazione, sembra che lo sviluppo del linguaggio andrà via via migliorando negli anni avvenire. Praticamente tutti i bambini con la sindrome alla fine impareranno a camminare. Quelli con il fenotipo lieve staranno seduti intorno all’anno e cammineranno intorno ai due anni, mentre la maggior parte dei pazienti con fenotipo classico staranno seduti prima dei due anni e cammineranno prima dei tre anni. Una minoranza dei bambini con la forma classica pronuncerà qualche parola, mentre la maggioranza di quelli con la forma lieve sarà in grado di parlare a frasi. I disturbi del comportamento sono comuni nella sindrome di Cornelia de Lange e sono più frequenti nei pazienti con grave ritardo mentale. E importante il ruolo delle cause mediche, come il reflusso gastroesofageo, quando si ha un peggioramento del comportamento: drammatici miglioramenti del comportamento non sono rari dopo terapia antireflusso. La prognosi, per quanto riguarda la gestione dei problemi di comportamento, e buona se viene individuato e trattato un problema clinico sottostante o se il problema di base era una difficolta di comunicazione che viene risolta con l’aiuto del linguaggio dei segni. In assenza di problemi di questo tipo, il comportamento rimane comunque un aspetto di difficile gestione da parte dei familiari e dei terapeuti.
Orecchie e udito
Un certo grado di deficit dell’udito e presente nel 90% degli individui con la sindrome di Cornelia de Lange. Ora si e visto che e di origine neurosensoriale e che di solito e bilaterale. Otiti medie e malattie croniche dell’orecchio medio (‘glue ear’) che si sovrappongono sono comuni e possono essere difficili da diagnosticare perché il canale uditivo esterno, in questa sindrome, tende ad essere molto stretto. Gli ausili auricolari vengono meglio tollerati quando sono impiantati precocemente.
Anomalie degli arti
Le anomalie degli arti che si possono vedere nella sindrome di Cornelia de Lange variano dalla presenza di mani piccole con dita corte a riduzioni bilaterali con assenza di mani e avambracci. Anomalie minori della mani includono impianto prossimale dei pollici, solco palmare singolo e clinodattilia del quinto dito. Più di rado, si possono vedere fusione di due dita o polidattilia e vi e una descrizione di un dito con due unghie. A livello dell’arto superiore, omero, ulna e radio sono di lunghezza ridotta, e occasionalmente il radio o l’ulna sono aplastici. E comune la brevità dei metacarpi, in particolare del primo. Sono comuni contratture in flessione dei gomiti, secondarie alla deformazione della metafisi prossimale del radio, che solitamente e sublussato.
Leggi anche: Cos’è un gene ed a che serve?
Problemi gastrointestinali
Il reflusso gastroesofageo rappresenta il più comune problema gastrointestinale, nella sindrome di Cornelia de Lange, e colpisce sia la forma classica sia quella lieve. Se non trattato, il reflusso può condurre a morbilità e mortalità significative. E comune che si sviluppi esofago di Barrett e stenosi esofagea, e si può sviluppare anche adenocarcinoma dell’esofago. La condizione si può presentare in individui con deficit di crescita, ridotta velocita di accrescimento, anemia, polmoniti ricorrenti, apnea o disturbi del comportamento. Poiché l’associazione tra reflusso e sindrome di Cornelia de Lange e emersa solo in tempi relativamente recenti, spesso al momento della diagnosi il danno esofageo e già grave. Malformazioni del tratto gastrointestinale che si presentino subito dopo la nascita includono stenosi pilorica in circa l’1% dei pazienti effetti o, molto raramente, un pancreas anulare che si manifesta con ostruzione duodenale.
Problemi cardiovascolari
Nel 20-25% dei pazienti con la forma classica della sindrome di Cornelia de Lange sono presenti difetti cardiaci congeniti, che sono invece rari nella forma lieve. L’anomalia più comune e la stenosi della valvola polmonare, sia isolata, sia in combinazione con un difetto del setto interventricolare. Una grande varietà di altri difetti e stata descritta in questa sindrome, compresa la tetralogia di Fallot, la coartazione aortica, il difetto del setto interatriale, il cuore sinistro ipoplasico, il ventricolo unico e il difetto del canale atrioventricolare. Nel 2% dei pazienti con la forma lieve si può osservare una modesta stenosi del ramo polmonare, che raramente richiede trattamento. I difetti cardiaci più gravi spesso si vedono nei pazienti con ernia diaframmatica e grave accorciamento degli arti. I bambini con difetti cardiaci più complessi di solito stentano a crescere e muoiono prima o durante l’intervento di chirurgia correttiva. Quelli con difetti più semplice, invece, hanno una buona prognosi.
Anomalie craniofacciali
Gli individui con la sindrome di Cornelia de Lange di frequente hanno il palato arcuato e alto. L’incidenza della palatoschisi varia tra il 18 e il 59% a seconda delle descrizioni. Al contrario di tante altre anomalie, la palatoschisi si può osservare anche nei pazienti con la forma lieve della sindrome. La palatoschisi non e mai associata a labioschisi. La prognosi di solito e buona, anche se i bambini con la forma lieve della sindrome di Cornelia de Lange e palatoschisi hanno meno probabilità di imparare a parlare rispetto a quelli senza.
Problemi oculistici
La ptosi della palpebra superiore e un reperto frequente specialmente nella forma classica, in cui può variare da un semplice problema estetico ad una interruzione dell’asse visivo. Anche la miopia e molto comune, ed e stata descritta nel 60% dei pazienti con la forma classica della sindrome di Cornelia de Lange. Sono molto comuni anche sintomi suggestivi di ostruzione del canale nasolacrimale, come gli occhi bagnati e le congiuntiviti ricorrenti. Questi sintomi, comunque, si risolvono quasi sempre spontaneamente col tempo, e sono rari dopo i sei anni.
Problemi al tratto genitourinario
La più frequente anomalia del tratto renale, in questa sindrome, e il rene a ferro di cavallo. Nei maschi, reperto estremamente comune e il criptorchidismo, che puo presentarsi in associazione con ipospadia e scroto ipoplasico. Nelle femmine, l’anomalia piu comune e l’utero bicorne.
Problemi neurologici
Sono state descritte molte anomalie del sistema nervoso centrale, ma nella maggior parte dei casi si tratta di pubblicazioni di singoli casi. Le convulsioni sono infrequenti, e si controllano bene con i farmaci.
CAUSE DELLA SINDROME DI CORNELIA DE LANGE
La sindrome di Cornelia de Lange è una malattia genetica, causata cioè da un’alterazione molto piccola nel patrimonio genetico della persona affetta. Nel maggio del 2004 è stato dimostrato che un’anomalia di una singola informazione genetica (gene NIPBL) è responsabile di tutte le manifestazioni cliniche nel 50% circa dei soggetti con questa condizione. La ricerca è ora impegnata a capire quale sia il difetto genetico di base nella restante parte delle persone con CDLS. L’alterazione genetica che causa la sindrome insorge solitamente in modo casuale nel patrimonio genetico del soggetto affetto; questo fa sì, quindi, che il rischio di ripetizione della malattia in altri figli dopo la nascita di un figlio affetto sia, di regola, molto basso. Esistono però rarissimi casi (una decina circa al mondo) in cui si è verificata una ripetizione della malattia; è per altro importante che ogni coppia che ha avuto un bambino affetto esegua un’appropriata consulenza genetica.
DIAGNOSI DELLA SINDROME DI CORNELIA DE LANGE
La diagnosi di CDLS è ancora prevalentemente basata sul riconoscimento delle caratteristiche cliniche da parte di un medico esperto (pediatra, genetista, ecc). Essa può essere fatta già alla nascita o nei mesi o negli anni successivi. Le recenti scoperte fanno sì che entro breve sarà possibile confermare la diagnosi clinica con l’esecuzione di un esame di laboratorio specifico (test genetico). Recentemente si è dimostrato anche che esistono pazienti con un quadro clinico meno marcato (forme mild = lievi) per i quali la diagnosi frequentemente è fatta più tardi e la prognosi è migliore.
Diagnosi prenatale e prevenzione
Poiché non vi sono test diagnostici per la sindrome, le gravidanze successive alla nascita di un bambino affetto vanno seguite con ecografie accurate, cercando anomalie strutturali come un igroma cistico, difetti a carico degli arti nel senso di una riduzione, un’ernia diaframmatica, anomalie cardiache o il caratteristico profilo della faccia. Il profilo del volto presenta una grave micrognatia con filtro lungo e sporgente, e naso piccolo. Il ritardo di crescita intrauterino, presente alla nascita nella maggior parte dei casi, raramente si vede prima del terzo trimestre. Nel 1983, Westergaard e colleghi descrissero la completa assenza della proteina-A plasmatica associata alla gravidanza (PAPP-A) in una serie di campioni di siero di donne gravide che che successivamente ebbero un bambino con la sindrome di Cornelia de Lange. In uno studio recente questo autore trovo che la PAPP-A era presente nei 19 campioni di siero materni analizzati, al secondo trimestre di gravidanza, ma che i suoi livelli erano significativamente ridotti. Sebbene non sia un test diagnostico, la PAPP-A può dimostrarsi un prezioso marcatore aggiuntivo per le gravidanze a rischio di ricorrenza o per quelle in cui siano state riscontrate delle anomalie compatibili con la sindrome di Cornelia de Lange.
Sospetti e conferme
La diagnosi prenatale della sindrome di Cornelia de Lange può essere sospettata dopo il precoce riscontro di un ritardo di crescita simmetrico, che diventa particolarmente evidente dopo la 25 settimana di gravidanza. La diagnosi può essere ulteriormente confermata quando si ricerchino segni ecografici aggiuntivi, come la facies tipica, difetti agli arti superiori, che di solito sono unilaterali, e varie anomalie viscerali tra cui l’ernia diaframmatica, la stenosi pilorica, il reflusso ureterale e lesioni cardiache di vario tipo (difetti del setto interatriale e interventricolare, stenosi dell’arteria polmonare o aortica, tetralogia di Fallot, finestra aorto-polmonare, canale atrioventricolare e ventricolo unico).
Il cariotipo fetale rappresenta il primo passo nella diagnosi differenziale in epoca prenatale, per escludere la trisomia 18 e la tetrasomia 12p, ed e essenziale, vista la somiglianza della sindrome di Cornelia de Lange con la duplicazione 3q. Nella maggior parte dei casi, la sindrome di Cornelia de Lange mostra un grave ritardo di crescita, oligodattilia, pollici in posizione prossimale e un filtro sporgente, mentre la duplicazione 3q e più spesso associata a craniosinostosi, palatoschisi e anomalie del tratto urinario.
PROBLEMI DEI PAZIENTI CON SINDROME DI CORNELIA DE LANGE
I problemi principali per i pazienti affetti da sindrome di Cornelia de Lange riguardano la scarsa crescita (esistono in proposito curve di crescita specifiche) e il ritardo psicomotorio e intellettivo. A questo proposito è importante che vengano intrapresi programmi di stimolazione precoci soprattutto per quanto riguarda l’area della comunicazione. Tra i problemi pediatrici più frequentemente associati vi sono il reflusso gastroesofageo e le sue complicanze (70-80% dei casi), le convulsioni (20% circa dei casi), difficoltà d’udito, otiti e sinusiti frequenti, blefariti e miopia. Possono poi insorgere problemi comportamentali (irritabilità, iperattività ecc.), a volte legati al mancato riconoscimento e trattamento di problemi medici in grado di causare dolore. La sopravvivenza di una persona affetta, che non presenti malformazioni maggiori gravi, non è particolarmente ridotta: oggi, infatti, sono ampiamente noti soggetti che hanno raggiunto l’età adulta.
TERAPIA NEI PAZIENTI CON SINDROME DI CORNELIA DE LANGE
Crescita e alimentazione
Se il bambino non è in grado di succhiare dal biberon, un introito calorico adeguato dovrebbe essere garantito per mezzo di nutrizione per via nasogastrica. Nei casi con grave reflusso gastroesofageo e difficolta di alimentazione, può essere necessario il posizionamento di un tubo per gastrostomia nel momento della funduplicatio.
Sviluppo e comportamento
Nei pazienti con la forma lieve della sindrome dovrebbe essere intrapresa terapia del linguaggio. Come aiuto alla comunicazione, dovrebbero essere introdotti il linguaggio dei segni o altre tecniche. Le patologie dell’orecchio medio dovrebbero essere trattate con antibiotici e, se necessario, con cannule di ventilazione. Ogni problema medico sottostante che deteriori il comportamento dovrebbe essere trattato. I problemi comportamentali di lunga data sono difficili da modificare, ma si possono migliorare con la modificazione comportamentale, guidata da uno psicologo del comportamento o da un pediatra dello sviluppo.
Anomalie degli arti
Non vi sono pubblicazioni che documentino che un approccio chirurgico alle anomalie degli arti possa dare dei benefici; anche i pazienti con grave ritardo mentale spesso hanno un controllo della motricità fine straordinariamente buono.
Problemi gastrointestinali
Nella pratica, sono molto pochi i pazienti che rispondono ad un trattamento conservativo del reflusso gastroesofageo, e la maggior parte necessita di un approccio chirurgico. Il trattamento iniziale del reflusso dovrebbe consistere nel suggerimento di assumere pasti piccoli e solidi e di rimanere in posizione eretta il più possibile. Va tentata la terapia con antiacidi, anti-H2 e inibitori della pompa protonica. Di solito si rende necessaria la funduplicatio, talvolta con inserzione di un tubo da gastrostomia.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
- Cosa sono gli alleli ed a che servono?
- Differenza tra cellule eucariote e procariote
- Virus e virioni: cosa sono, come sono fatti, come funzionano e come si riproducono
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
Condividi questo articolo:

 Jason Padgett, che vedete sorridente nella foto qui sopra, era una persona che amava divertirsi. Gonfiare i bicipiti, scolpirsi i capelli col gel, fare tardi nei bar. Poi improvvisamente tutto è cambiato. Nella mente di questo trentunenne americano allergico ai libri si è acceso un sesto senso per la matematica. E’ diventato una sorta di “idiota sapiente” anche se, visto che lui “savant” non ci è nato, ma ci è diventato, sarebbe più corretto parlare di “sindrome del savantismo acquisito“. In letteratura scientifica sono davvero pochi i casi come quello di Jason: si calcola che in tutto il mondo esistano solo circa 40 persone che sono state colpite da tale particolarissima sindrome. Nel caso di Jason il verbo colpire è particolarmente azzeccato: l’evento che lo ha trasformato è stata una serie di pugni alla testa sferrati da un paio di sconosciuti in un locale notturno. Il ragazzo è caduto a terra, ha perso i sensi e quando si è ripreso ogni cosa aveva iniziato a splendere di una luce strana.
Jason Padgett, che vedete sorridente nella foto qui sopra, era una persona che amava divertirsi. Gonfiare i bicipiti, scolpirsi i capelli col gel, fare tardi nei bar. Poi improvvisamente tutto è cambiato. Nella mente di questo trentunenne americano allergico ai libri si è acceso un sesto senso per la matematica. E’ diventato una sorta di “idiota sapiente” anche se, visto che lui “savant” non ci è nato, ma ci è diventato, sarebbe più corretto parlare di “sindrome del savantismo acquisito“. In letteratura scientifica sono davvero pochi i casi come quello di Jason: si calcola che in tutto il mondo esistano solo circa 40 persone che sono state colpite da tale particolarissima sindrome. Nel caso di Jason il verbo colpire è particolarmente azzeccato: l’evento che lo ha trasformato è stata una serie di pugni alla testa sferrati da un paio di sconosciuti in un locale notturno. Il ragazzo è caduto a terra, ha perso i sensi e quando si è ripreso ogni cosa aveva iniziato a splendere di una luce strana.









 “Fa’ che il cibo sia la tua medicina e che la medicina sia il tuo cibo”
“Fa’ che il cibo sia la tua medicina e che la medicina sia il tuo cibo” Che siamo fatti di atomi credo lo sappiate già tutti; ma lo sapete che gli atomi sono fatti al 99,9999999999% di vuoto? Se togliessimo tutto lo spazio vuoto tra nucleo ed elettroni, tutti i 7 miliardi di abitanti della terra entrerebbero nello spazio di una palla da tennis! Ma se siamo fatti di atomi, e gli atomi sono fatti quasi solamente di vuoto, come mai se ci sediamo sulla sedia non cadiamo letteralmente per terra? Cominciamo dal principio!
Che siamo fatti di atomi credo lo sappiate già tutti; ma lo sapete che gli atomi sono fatti al 99,9999999999% di vuoto? Se togliessimo tutto lo spazio vuoto tra nucleo ed elettroni, tutti i 7 miliardi di abitanti della terra entrerebbero nello spazio di una palla da tennis! Ma se siamo fatti di atomi, e gli atomi sono fatti quasi solamente di vuoto, come mai se ci sediamo sulla sedia non cadiamo letteralmente per terra? Cominciamo dal principio!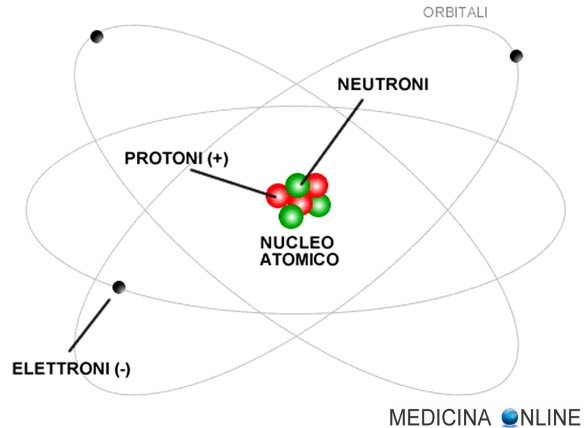
 Io, sinceramente, no; la vita perderebbe il suo lato più terrorizzante ma che è anche il lato più bello: l’imprevedibilità! E voi che ne pensate? Viene chiamato il “test della morte“, ed è un esame del sangue che permetterebbe di predire le possibilità che una persona, anche apparentemente non malata, possa morire per qualche problema di salute entro i prossimi cinque anni. A metterlo a punto è stato un gruppo di scienziati estoni e finlandesi: intervistati dal quotidiano inglese Telegraph, hanno dichiarato che si tratta di una scoperta eccezionale perché un semplice esame del sangue può prevedere future possibilità di morte.
Io, sinceramente, no; la vita perderebbe il suo lato più terrorizzante ma che è anche il lato più bello: l’imprevedibilità! E voi che ne pensate? Viene chiamato il “test della morte“, ed è un esame del sangue che permetterebbe di predire le possibilità che una persona, anche apparentemente non malata, possa morire per qualche problema di salute entro i prossimi cinque anni. A metterlo a punto è stato un gruppo di scienziati estoni e finlandesi: intervistati dal quotidiano inglese Telegraph, hanno dichiarato che si tratta di una scoperta eccezionale perché un semplice esame del sangue può prevedere future possibilità di morte.

 Immaginate di soffrire il mal di denti ma di non provare alcun dolore. Oppure di ricevere un pugno sul naso e continuare senza problemi a fare quel che stavate facendo.
Immaginate di soffrire il mal di denti ma di non provare alcun dolore. Oppure di ricevere un pugno sul naso e continuare senza problemi a fare quel che stavate facendo.
