
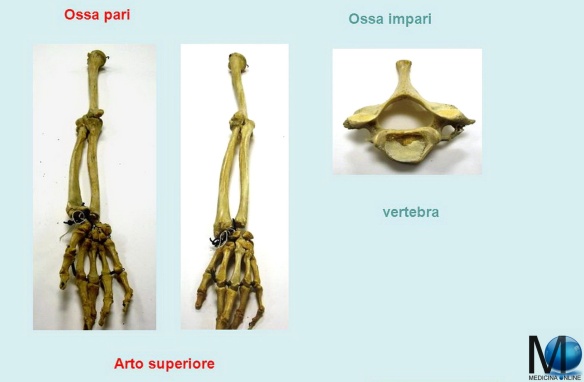
 Le ossa pari (anche chiamate “ossa simmetriche”) sono quelle che nel nostro organismo sono doppie, presenti cioè in due copie. Sono ossa pari ad esempio il femore, le costole, l’omero ed il radio, di cui esistono una copia appartenente al lato destro del corpo, ed un’altra al lato sinistro.
Le ossa pari (anche chiamate “ossa simmetriche”) sono quelle che nel nostro organismo sono doppie, presenti cioè in due copie. Sono ossa pari ad esempio il femore, le costole, l’omero ed il radio, di cui esistono una copia appartenente al lato destro del corpo, ed un’altra al lato sinistro.
Le ossa impari (o “asimmetriche”) sono quelle contenute in una sola copia nel corpo umano, e sono presenti nella linea mediana del corpo (un’asse immaginaria che attraversa longitudinalmente la colonna vertebrale) per questo prendono anche il nome di ossa mediane. Esempi di ossa impari sono: l’osso frontale (la fronte), l’osso occipitale (che si trova alla base del cranio, vicino al collo), le vertebre (che compongono la colonna vertebrale) oppure lo sfenoide, osso all’interno del cranio.
Leggi anche:
- Differenza tra osso compatto e spugnoso
- Differenza tra osso fibroso e lamellare
- I tipi di tessuto osseo: cellule, matrice, formazione e struttura
- Differenza tra osso e cartilagine: funzioni, composizione e durezza
- Differenza tra osso sacro e coccige
- Osso sacro e coccige: dove si trovano ed a che servono?
- Differenza ossa umane e animali
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- A cosa serve il midollo osseo?
- Differenza tra midollo giallo e rosso
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Differenza tra osso ed avorio: come distinguerli
- Quante ossa ed articolazioni abbiamo nel nostro corpo?
- Quanto pesano scheletro ed ossa?
- Quante ossa ha il piede e come si chiamano?
- Differenza tra costole e vertebre
- Differenza tra costola incrinata e rotta
- Frattura costale multipla, volet costale e pneumotorace
- Cos’è una costa? Differenza tra costole e coste
- Gabbia toracica: dove si trova, a che serve e da cosa è composta
- Costola rotta (frattura costale): sintomi, diagnosi e terapia
- Costola incrinata: sintomi, terapia e tempi di recupero
- Non sono grasso: ho la costituzione robusta e le ossa grosse
- Differenza tra legamento e tendine con esempi
- Si rompe il femore a 109 anni: “Ora voglio andare a bere il caffè al bar”
- Differenza tra mascella e mandibola: sono sinonimi?
- Quante ossa ci sono nella mano e come si chiamano?
- Come si chiamano le dita dei piedi?
- Differenza tra frattura composta, composta, esposta e patologica
- Callo osseo e pseudoartrosi, quando la frattura non guarisce: cause, diagnosi e terapie
- Differenza tra osteoblasti, osteoclasti ed osteociti
- Di cos’è fatto un osso, a che serve e perché è così resistente?
- Articolazione del ginocchio: com’è fatta, quali sono le patologie, i sintomi e gli esami da fare ?
- Mastoidite, cos’è e dove si trova l’osso mastoide?
- Differenza tra pube ed osso iliaco: anatomia e funzioni
- Legamenti: cosa sono, dove si trovano ed a che servono?
- Differenza tra legamento ed articolazione con esempi
- Osteoporosi: cos’è la Mineralometria Ossea Computerizzata (MOC), a cosa serve, come si interpretano i risultati?
- Osteoporosi: cause, diagnosi e cure
- A che serve l’osso ioide e dove si trova? Cos’è il pomo d’Adamo?
- Mandibola GH, ormone della crescita e doping nello sport
- Ormone della crescita (GH) a che serve e da cosa è prodotto?
- Ormone della crescita (GH): body building e doping in palestra
- Gomito del tennista (epicondilite): cos’è, quanto dura e rimedi
- Ti piace scrocchiare le dita? Ecco cosa succede alle tue ossa e i danni che rischi
- Differenza tra ossa lunghe, corte, irregolari e piatte con esempi
- Frattura della spalla e dell’omero prossimale: sintomi e cure
- Che differenza c’è tra la risonanza magnetica, la TAC, la PET, la radiografia, l’ecografia e l’endoscopia?
- L’altezza media italiana nel 2017 di maschi e femmine
- L’altezza media italiana 2017 da 1 a 19 anni di maschi e femmine
- L’altezza media mondiale nel 2017 di maschi e femmine [TABELLA]
- Altezza: quando si può parlare di nanismo o gigantismo
- Quanto è alto l’uomo più alto del mondo?
- Quanto è alto l’uomo più basso del mondo?
- Articolazioni mobili, semimobili, sinoviali e fisse: struttura e funzioni
- Dismetria degli arti inferiori: una gamba è più corta dell’altra
- Femore: anatomia, funzioni e muscoli in sintesi
- Quante ossa ci sono nella mano e come si chiamano?
- Quante ossa ha il piede e come si chiamano?
- Cosa sono e qual è la differenza tra massa magra e massa grassa? Tutte le percentuali di grasso, ossa e muscoli
- Quanti litri e percentuale di acqua sono presenti nel nostro corpo?
- Differenza tra femore e anca
- Differenza tra frattura composta, composta, esposta e patologica
- Femore rotto: tipi di frattura, sintomi, intervento, riabilitazione e conseguenze
- Artrosi: il latte fa bene alle ginocchia (delle donne)
- Allattamento materno, artificiale e misto: quale latte per il bambino?
- Il latte fa male alle ossa? Si, ma solo se sei iscritto a Facebook
- Artrosi dell’anca: cause, sintomi, prevenzione e trattamento
- Differenze tra artrite ed artrosi: sintomi comuni e diversi
- Sindrome del tunnel carpale: prevenzione, diagnosi e cura di una dolorosa patologia
- Crollo vertebrale nell’anziano da osteoporosi e tumore: sintomi, diagnosi e terapia
- Malattie reumatiche: cosa sono, come si curano, sono pericolose?
- Differenza artrite reumatoide e artrite psoriasica: sintomi comuni e diversi
- Artrite psoriasica e spondiloartriti sieronegative: sintomi, diagnosi e cura
- Fattore reumatoide alto o basso? Valori normali e Reuma test
- Sindrome del piriforme: sintomi, esercizi, cura e recupero
- Osso omero: anatomia e funzioni in sintesi
- Scapola: dove si trova ed a che serve?
- Terapia con Infrarossi per il dolore
- Fibromialgia: dove si trovano i tender points che provocano dolore alla palpazione?
- Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
- Lupus eritematoso sistemico (LES): cause, sintomi e terapie
- Come viene effettuata una ecografia articolare (muscolo tendinea) ed a cosa serve?
- Differenza tra distorsione, lussazione, sublussazione e strappo muscolare
- Quanti muscoli abbiamo nel nostro corpo?
- Pubalgia in gravidanza: cause e rimedi del dolore all’osso pubico
- Pubalgia acuta e cronica: sintomi, esercizi e rimedi
- Differenza tra pube e inguine
- Differenza tra muscoli volontari, involontari, scheletrici e viscerali
- I muscoli: come sono fatti, come funzionano e cosa rischiano quando ti alleni
- Differenze tra muscolo striato, scheletrico, liscio, cardiaco, superficiale e profondo
- Quanto è alto l’uomo più alto del mondo?
- Quanto è alto l’uomo più basso del mondo?
- Nanismo: sintomi, cura, cause, terapia, diagnosi e prevenzione
- Charlotte, la bambina più piccola del mondo
- Quando essere troppo alti è una malattia: il gigantismo
- Differenza tra gigantismo ed acromegalia
- Aumentare l’altezza è possibile? Esercizi e trucchi
- Lo sai che la mattina sei alto due cm in più rispetto alla sera? Tutto quello che vorresti sapere sull’altezza e su come crescere di più
- Hai una costituzione corporea robusta o esile? Impara a valutarlo scientificamente
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
 Il bio-regolatore “per eccellenza” dello sviluppo osseo è la somatotropina, meglio conosciuta con il nome di ormone della crescita (GH). Esso viene secreto in maniera “pulsatile”, è responsabile dello stimolo anabolico di vari tessuti e, sia nei maschi che nelle femmine, la sua produzione (da parte dell’ipofisi o ghiandola pituitaria) dipende dalla regolazione ipotalamica e risulta sensibilmente maggiore durante il periodo dello sviluppo. In età adulta la produzione di somatotropina si riduce e (rispettando i ritmi circadiani) raggiunge il vertice di concentrazione plasmatica nelle ore notturne. Il GH interviene positivamente sulla crescita dell’osso (quindi anche dell’altezza) SOLO durante il periodo dello sviluppo, durante il quale può essere utilizzato sotto stretta supervisione medica per correggere eventuali gravi deficit di sviluppo. In età adulta, invece, qualsiasi tentativo di favorire la produzione fisiologica di GH (vedi integrazione di arginina) o di incrementarne i livelli plasmatici attraverso iniezioni esogene (come nel doping), oltre ad essere PERICOLOSO, risulta del tutto inutile nell’aumento dell’altezza. Per approfondire l’argomento, leggi:
Il bio-regolatore “per eccellenza” dello sviluppo osseo è la somatotropina, meglio conosciuta con il nome di ormone della crescita (GH). Esso viene secreto in maniera “pulsatile”, è responsabile dello stimolo anabolico di vari tessuti e, sia nei maschi che nelle femmine, la sua produzione (da parte dell’ipofisi o ghiandola pituitaria) dipende dalla regolazione ipotalamica e risulta sensibilmente maggiore durante il periodo dello sviluppo. In età adulta la produzione di somatotropina si riduce e (rispettando i ritmi circadiani) raggiunge il vertice di concentrazione plasmatica nelle ore notturne. Il GH interviene positivamente sulla crescita dell’osso (quindi anche dell’altezza) SOLO durante il periodo dello sviluppo, durante il quale può essere utilizzato sotto stretta supervisione medica per correggere eventuali gravi deficit di sviluppo. In età adulta, invece, qualsiasi tentativo di favorire la produzione fisiologica di GH (vedi integrazione di arginina) o di incrementarne i livelli plasmatici attraverso iniezioni esogene (come nel doping), oltre ad essere PERICOLOSO, risulta del tutto inutile nell’aumento dell’altezza. Per approfondire l’argomento, leggi: 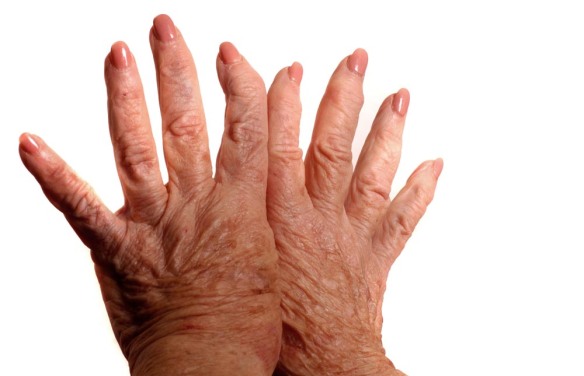 L’artrite reumatoide è una poliartrite infiammatoria cronica, anchilosante e progressiva a patogenesi autoimmunitaria e di eziologia sconosciuta, principalmente a carico delle articolazioni sinoviali. Può provocare deformazione e dolore che possono portare fino alla perdita della funzionalità articolare.
L’artrite reumatoide è una poliartrite infiammatoria cronica, anchilosante e progressiva a patogenesi autoimmunitaria e di eziologia sconosciuta, principalmente a carico delle articolazioni sinoviali. Può provocare deformazione e dolore che possono portare fino alla perdita della funzionalità articolare. Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard.
Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard. Ecco in questa tabella, il range di altezza media degli italiani in bambini e giovani da uno a 19 anni:
Ecco in questa tabella, il range di altezza media degli italiani in bambini e giovani da uno a 19 anni: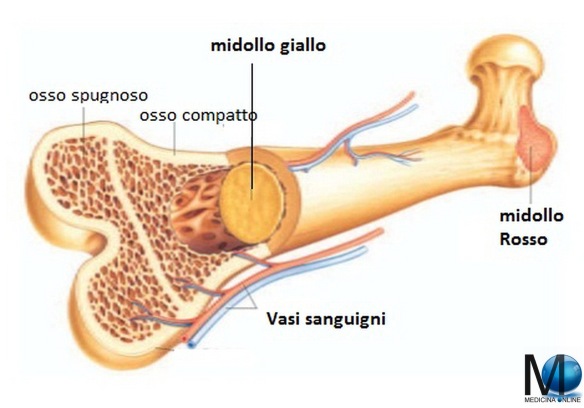 Qual è la differenza tra il midollo osseo e le cellule staminali?
Qual è la differenza tra il midollo osseo e le cellule staminali?