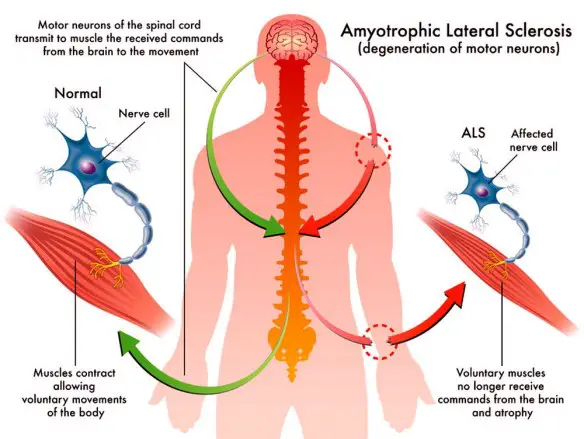
 La sclerosi multipla, SM, e la sclerosi laterale amiotrofica, SLA, vengono confuse molto spesso tra i non “addetti ai lavori”, questo a causa del termine “sclerosi” che le due patologie hanno in comune, pur essendo molto diverse tra loro.
La sclerosi multipla, SM, e la sclerosi laterale amiotrofica, SLA, vengono confuse molto spesso tra i non “addetti ai lavori”, questo a causa del termine “sclerosi” che le due patologie hanno in comune, pur essendo molto diverse tra loro.
Sclerosi laterale amiotrofica e sclerosi multipla: caratteristiche comuni
Pur essendo molto diverse tra loro, sclerosi laterale amiotrofica e sclerosi multipla hanno però in comune alcune caratteristiche:
- sono malattie che interessano il sistema nervoso;
- sono croniche;
- determinano disturbi neuromotori;
- degenerano progressivamente nel tempo;
- non possono essere curate: le terapie servono solo per ridurre i sintomi e rallentare la loro progressione.
Leggi anche: Morbo di Parkinson: cause, sintomi, decorso, terapie
Differenze nell’età e nell’incidenza
La sclerosi laterale amiotrofica interessa principalmente gli adulti over 50, in particolare tra i 50 e i 70 anni, con una leggera prevalenza negli uomini rispetto alle donne e con un’incidenza generale decisamente inferiore rispetto alla sclerosi multipla. La sclerosi multipla, invece, viene anche detta la malattia invalidante del giovane adulto, in quanto riguarda prevalentemente soggetti tra i 20 e i 40 anni. In questo, soprattutto donne.
Leggi anche: Morbo di Alzheimer: cause, sintomi, decorso, terapie
Differenze nelle cause
Sia per quel riguarda la SLA che per la sclerosi multipla, le cause che determinano la loro insorgenza sono ancora oggi difficili da definire. Per quanto riguarda la SLA si sa, però, che è una malattia multifattoriale, dovuta a cause diverse e concomitanti. Ad esempio carenza di nutrienti per le cellule del sistema nervoso, eccesso di anticorpi, virus. Una buona dose di responsabilità è da attribuire all’ereditarietà e alla predisposizione genetica, a differenza della sclerosi multipla per cui l’elemento ereditario ha minor impatto sull’insorgenza della malattia. Influiscono invece fattori ambientali e geografici: la sclerosi multipla si manifesta soprattutto in luoghi lontani dall’equatore. Le persone che vivono molto più vicino all’equatore sono esposte ad una grande quantità di luce solare per tutto l’anno e per questo tendono ad avere dei livelli più alti di vitamina D che previene l’insorgenza della sclerosi multipla.
Le zone colpite e le conseguenze sono diverse
La sclerosi multipla intacca la mielina, che garantisce la corretta conduzione degli stimoli nervosi. Non a caso la SM è soprannominata malattia demielinizzante, proprio perché provoca la perdita di mielina, da cui dipendono i danni neurologici e il rallentamento dell’impulso nervoso. Il nome sclerosi si deve alla formazione di cicatrici in varie zone mentre il termine Multipla si riferisce al fatto che la malattia può intaccare le diverse parti in tempi diversi. Le zone colpite dalle cicatrici che si vanno formando sono:
- il sistema nervoso centrale;
- il cervello;
- il midollo spinale.
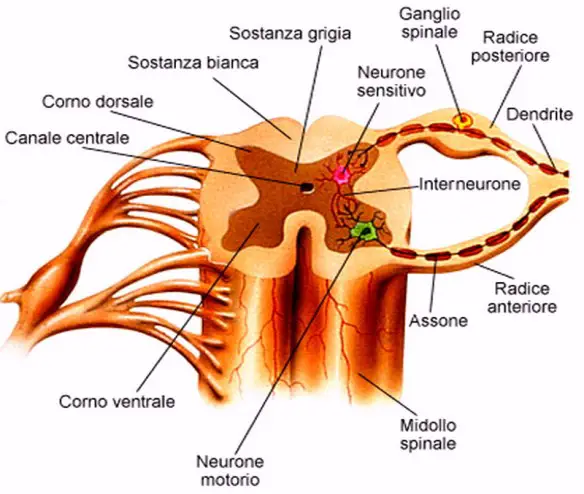
La SLA, anche detta “malattia dei motoneuroni“, intacca invece solo i motoneuroni, cellule adibite al controllo del movimento muscolare, che progressivamente muoiono. Anche in questo caso la malattia provoca la formazione di cicatrici che però indeboliscono solo i muscoli, localizzate nei cordoni laterali del midollo spinale. Ne consegue che il soggetto colpito dalla SLA perde progressivamente forza muscolare e in alcuni casi l’indebolimento può addirittura sfociare in paralisi. I motoneuroni colpiti possono essere di due tipologie:
- I motoneurone, detto anche centrale o corticale, localizzato nella corteccia cerebrale, adibito al trasporto del segnale dal cervello al midollo spinale
- II motoneurone, detto anche periferico o spinale, localizzato nel midollo spinale, adibito al trasporto del segnale in periferia ai muscoli.
Leggi anche: Differenza tra morbo di Alzheimer e morbo di Parkinson: sintomi comuni e diversi
Le diagnosi sono diverse
La sclerosi mutipla viene solitamente diagnosticata valutando i vari episodi di disturbi neurologici disseminati nel tempo, che possono fornire una mappa per ricostruire il quadro clinico del paziente. Sebbene sia caratterizzata anche da alcuni sintomi tipici, essi spesso non sono sufficienti a permetterne l’individuazione precisa. Tuttavia esistono anche alcuni esami mirati, come la RM e l’analisi del liquido cerebrospinale, che permettono di diagnosticarla con maggiore sicurezza e facilità. La diagnosi di SLA è invece più difficile perché questa patologia non comporta sintomi specifici, quindi si va spesso per esclusione dopo aver effettuato un esame neurologico per individuare eventuali lesioni del I e II motoneurone associato ad altri esami strumentali.
I sintomi sono diversi
Per quanto riguarda la sclerosi multipla si possono individuare alcuni sintomi e segni tipici, sebbene ciò che aiuta a individuare la patologia sia più che altro la storia clinica del paziente, attraverso l’individuazione di disturbi neurologici disseminati nel tempo e localizzati nelle zone caratteristiche. I sintomi e segni più comuni della SM sono:
- disturbi motori più o meno gravi;
- formicolii;
- sensazione di punture;
- calo o sdoppiamento della vista;
- vertigini accompagnate spesso da vomito e nausea;
- instabilità, barcollamento, disturbi dell’equilibrio;
- disturbi intestinali;
- a volte disturbi urinari;
- a volte disturbi sessuali.
La sclerosi multipla può comunque determinare virtualmente qualsiasi sintomo e segno relativo al sistema nervoso. Può anche determinare patologie psichiatriche, come la despressione (sia in risposta alla notizia della diagnosi, sia a causa del danno del tessuto nervoso).
Per quanto riguarda la SLA, essa colpisce solo il sistema motorio provocando sintomi a livello muscolare. Tra i sintomi e segni più comuni, ricordiamo:
- debolezza sempre più intensa, che di solito parte dalle mani e dai piedi per poi intaccare il resto del corpo;
- rigidità;
- crampi muscolari;
- contrazioni involontarie.
Le terapie sono diverse
Nel caso della sclerosi multipla, ad oggi non esiste una cura definitiva: le terapie cercano di ridurre e prevenire le ricadute e rallentare il decorso della malattia. Esse includono principalmente la somministrazione di farmaci appositi, in particolare i farmaci antinfiammatori steroidei quali l’adrenocorticotropina (conosciuto come ACTH), il prednisone, il metilprednisolone, il prednisolone, il betametasone e il dexametasone. Durante gli attacchi sintomatici, la somministrazione di alte dosi di corticosteroidi per via endovenosa, come il metilprednisolone, è la terapia di routine per le recidive acute della malattia in forma recidivante-remittente, in quanto ha dimostrato efficacia nel ridurre la gravità e la durata delle esacerbazioni. Recentemente sono stati approvati alcuni i farmaci modificanti la malattia, tra cui: l’interferone beta-1a, l’interferone beta-1b, il glatiramer acetato, il mitoxantrone (un immunosoppressore usato anche in chemioterapia), il natalizumab (un anticorpo monoclonale umanizzato immunomodulatore che impedisce la migrazione delle cellule T dal torrente circolatorio al sistema nervoso centrale), il fingolimod e il teriflunomide, rispettivamente il primo e il secondo farmaco a somministrazione orale a essere disponibili. I trattamenti modificanti la malattia sono in grado di ridurre il tasso di progressione della malattia, ma non di arrestarla. Con la progressione della sclerosi multipla, la sua sintomatologia tende ad aumentare. La malattia è associata a una varietà di sintomi e deficit funzionali che si traducono in una serie di menomazioni e disabilità progressive. La gestione di questi deficit è quindi molto importante. Sia la terapia farmacologica che la neuroriabilitazione hanno dimostrato di poter alleviare alcuni sintomi, anche se non influenzano la progressione della malattia. Alcuni sintomi, come l’incontinenza urinaria e la spasticità, hanno una buona risposta ai farmaci, mentre la gestione di molti altri risulta più complessa. Le persone colpite da sclerosi multipla necessitano, inoltre, di una terapia rivolta alle eventuali malattie collaterali, alle infezioni delle vie urinarie e alle piaghe da decubito. Molto utili contro la spasticità degli arti si sono dimostrati i farmaci miorilassanti e la fisiochinesiterapia. Nell’ambito delle terapie sintomatiche, è possibile usare, a seconda del tipo di disturbi e della loro entità, farmaci per la spasticità, la fatica, le disfunzioni vescicali, i disturbi delle sensibilità e così via. Il farmaco di prima scelta nel trattamento della spasticità è il baclofen.
Per la SLA non esistono ancora farmaci specifici capaci di curare del tutto la patologia: l’unico farmaco approvato dalla FDA è il riluzolo, che agisce sui livelli di glutammato, la cui assunzione può rallentare la progressione della malattia. Test clinici in pazienti con SLA hanno mostrato che il riluzolo prolunga la sopravvivenza fino a soli tre mesi, e può estendere il tempo di sopravvivenza soprattutto nei pazienti con SLA ad inizio bulbare. Il farmaco estende anche il tempo durante il quale il paziente può rimanere libero dal supporto ventilatorio. Il riluzolo non può invertire il danno subito dai motoneuroni, ed i pazienti che prendono il farmaco devono essere monitorizzati per il danno epatico ed altri possibili effetti collaterali. Sono previsti trattamenti il cui scopo è migliorare la qualità della vita del paziente, riducendo il più possibile eventuali complicanze. Tra essi si annoverano:
- riabilitazione;
- ventilazione di supporto;
- somministrazione di appositi integratori alimentari.
Le cure palliative vengono fornite al meglio da team multidisciplinari costituiti da professionisti dell’assistenza come medici, farmacisti, fisioterapisti, terapisti occupazionali, e logopedisti; nutrizionisti; assistenti sociali; ed infermieri specializzati nell’assistenza domiciliare e negli hospice per lungodegenti. Lavorando con i pazienti ed il personale sanitario, questi “team di assistenza” possono pianificare un piano individualizzato di terapia medica e fisica e fornire apparecchiature speciali destinate a mantenere i pazienti nella migliore situazione di mobilità e comfort che si possa ragionevolmente raggiungere.
Le ricadute
Le ricadute nella sclerosi multipla, anche dette recidive, riacutizzazioni, peggioramenti, attacchi, consistono nella comparsa acuta o sub-acuta di anormalità neurologiche per 24 ore senza che compaiono febbre o infezioni di alcun tipo. Tra una ricaduta e l’altra possono trascorrere alcune settimane o addirittura anni a seconda dei casi e purtroppo non c’è modo di prevedere l’episodio nè di individuarne le cause, nonostante le tante ipotesi formulate dalle ricerche a tema. Tra queste si annoverano eventuali traumi, stress, infezioni e vaccinazioni,ma come premesso si tratta solo di supposizioni non confermate da dati certi. Sebbene le ricadute si guariscano spontaneamente, di solito al paziente vengono somministrati steroidi per pochi giorni in modo da ridurre al minimo la gravità e la durata. Diverso è il caso della SLA poiché questa malattia ha un decorso diverso rispetto alla SM: essa progredisce man mano che i motoneuroni muoiono nel corso di mesi o anni, in modo progressivo, senza le ricadute tipiche della sclerosi multipla. Nella SLA si verifica quindi un peggioramento progressivo, di pari passo alla morte dei motoneuroni.
Per approfondire:
Leggi anche:
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Demenza senile: cause, sintomi, decorso e cure
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Apparato respiratorio: anatomia in sintesi, struttura e funzioni
- Apparato digerente: cos’è, com’è fatto, a che serve e come funziona?
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- A cosa serve il midollo osseo?
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Coma da emorragia cerebrale: quanto può durare?
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski positivo: quali patologie può indicare?
- Segno di Babinski bilaterale: cos’è e che patologia indica?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Babinski ed alluce muto: cosa significa?
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Differenza tra emiparesi ed emiplegia
- Differenza tra emiplegia destra e sinistra
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Paraplegia: erezione, disfunzione erettile ed eiaculazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Differenza tra paraplegico e tetraplegico
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Differenza tra paralisi e paresi
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Si può morire di epilessia?
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
Condividi questo articolo:

 L’atrofia muscolare progressiva, detta anche atrofia muscolare di Duchenne-Aran o malattia di Duchenne-Aran, nota anche con l’acronimo AMP o in inglese PMA (Progressive muscular atrophy), è una malattia neurologica configurabile attualmente quale rara forma di malattia del motoneurone (Motor neurone disease, MND), considerata talvolta relativa o collegata alla sclerosi laterale amiotrofica. La PMA colpisce solo il 2° motoneurone, lasciando intatto il 1°.
L’atrofia muscolare progressiva, detta anche atrofia muscolare di Duchenne-Aran o malattia di Duchenne-Aran, nota anche con l’acronimo AMP o in inglese PMA (Progressive muscular atrophy), è una malattia neurologica configurabile attualmente quale rara forma di malattia del motoneurone (Motor neurone disease, MND), considerata talvolta relativa o collegata alla sclerosi laterale amiotrofica. La PMA colpisce solo il 2° motoneurone, lasciando intatto il 1°. La SLA, Sclerosi laterale amiotrofica, conosciuta anche come “malattia dei motoneuroni“, “malattia di Lou Gehrig” (dal nome del giocatore di baseball la cui malattia nel 1939 fu portata all’attenzione pubblica) o “malattia di Charcot” (dal cognome del neurologo francese che per la prima volta descrisse questa patologia nel 1860), è una malattia neurodegenerativa progressiva che colpisce i motoneuroni, ovvero le cellule nervose cerebrali e del midollo spinale in grado di regolare l’attività di contrazione dei muscoli volontari. La morte di queste particolari cellule avviene gradualmente, in un lasso di tempo che può andare da diversi mesi a diversi anni, e la gravità può variare molto da un paziente all’altro.
La SLA, Sclerosi laterale amiotrofica, conosciuta anche come “malattia dei motoneuroni“, “malattia di Lou Gehrig” (dal nome del giocatore di baseball la cui malattia nel 1939 fu portata all’attenzione pubblica) o “malattia di Charcot” (dal cognome del neurologo francese che per la prima volta descrisse questa patologia nel 1860), è una malattia neurodegenerativa progressiva che colpisce i motoneuroni, ovvero le cellule nervose cerebrali e del midollo spinale in grado di regolare l’attività di contrazione dei muscoli volontari. La morte di queste particolari cellule avviene gradualmente, in un lasso di tempo che può andare da diversi mesi a diversi anni, e la gravità può variare molto da un paziente all’altro.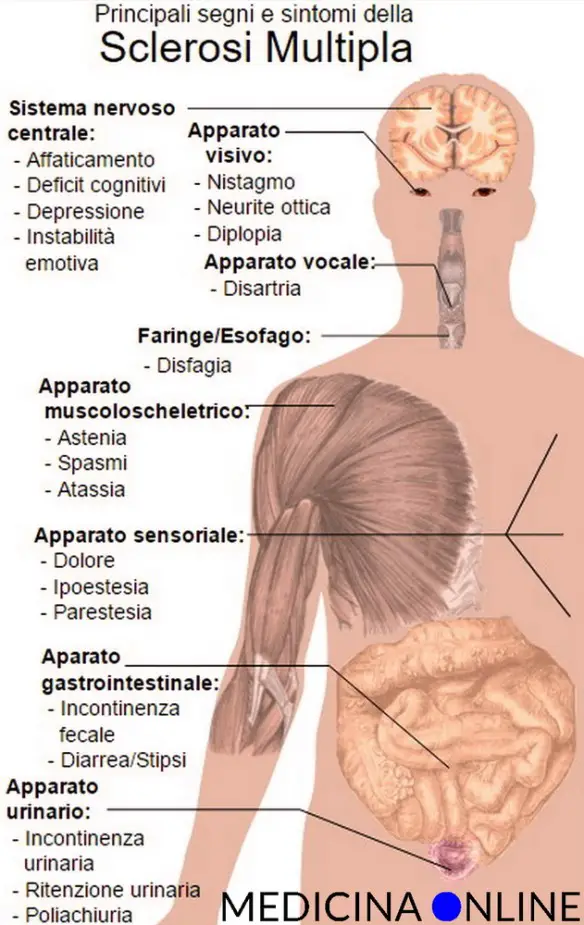
 Il decorso della malattia di Alzheimer è diviso in quattro fasi, contraddistinte da sintomi diversi:
Il decorso della malattia di Alzheimer è diviso in quattro fasi, contraddistinte da sintomi diversi: La scala di Hoehn e Yahr viene usata in campo medico per descrivere i sintomi della progressione del morbo di Parkinson. È stata originariamente pubblicata nel 1967 sulla rivista Neurology da Melvin Yahr e Margaret Hoehn e comprendeva gli stadi da 1 a 5. Da allora, è stata proposta una scala modificata, con l’aggiunta degli stadi 1,5 e 2,5 che descrivono il decorso intermedio della malattia.
La scala di Hoehn e Yahr viene usata in campo medico per descrivere i sintomi della progressione del morbo di Parkinson. È stata originariamente pubblicata nel 1967 sulla rivista Neurology da Melvin Yahr e Margaret Hoehn e comprendeva gli stadi da 1 a 5. Da allora, è stata proposta una scala modificata, con l’aggiunta degli stadi 1,5 e 2,5 che descrivono il decorso intermedio della malattia.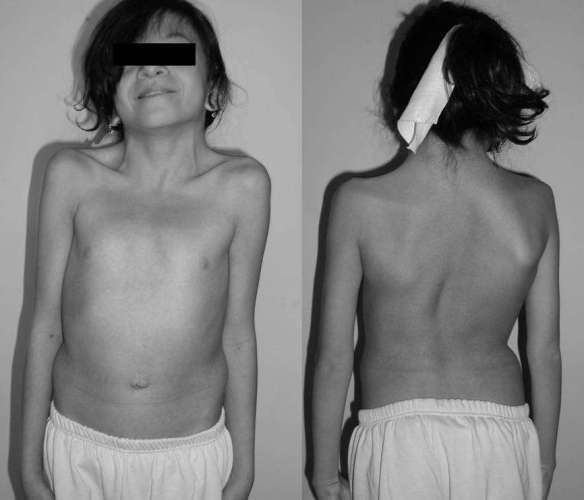 La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).
La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).