
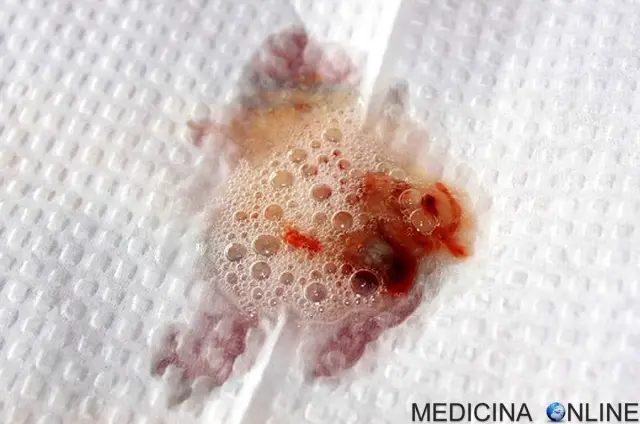
Espettorato con sangue
La fibrosi cistica è una rara malattia genetica autosomica recessiva causata da una mutazione di un gene sito sul cromosoma 7, il quale codifica per una proteina che funziona come canale per il cloro, detta CFTR (Cystic Fibrosis Transmembrane conductance Regulator). La fibrosi cistica, un tempo mortale nel primo anno di nascita, è oggi una malattia che può essere curata, tuttavia rimane una malattia fortemente debilitante, che abbassa la qualità della vita del paziente e diminuisce la sua aspettativa di vita.
Sintomi e segni
I segni distintivi della fibrosi cistica sono pelle salata, scarsa crescita e scarso aumento di peso nonostante una normale assunzione di cibo, accumulo di muco denso e appiccicoso, infezioni polmonari frequenti e tosse o mancanza di respiro. I maschi possono non essere fertili a causa dell’assenza congenita dei vasi deferenti. I sintomi spesso compaiono durante l’infanzia, come l’ostruzione intestinale a causa di ileo patologico da meconio nei neonati. Quando i bambini crescono, vi sono complicanze nel rilascio del muco negli alveoli. Le cellule ciliate epiteliali del paziente hanno una proteina mutata che porta alla produzione di muco anormalmente viscoso. La scarsa crescita nei bambini si presenta tipicamente come l’incapacità di aumentare di peso o di altezza rispetto ai loro coetanei. Spesso la condizione non viene diagnosticata fino a quando non si cercano le cause di questa scarsa crescita. Le cause della mancata crescita sono multifattoriali e comprendono l’infezione polmonare cronica, il cattivo assorbimento delle sostanze nutrienti attraverso il tratto gastrointestinale e l’aumento della domanda metabolica a causa dello stato cronico di malattia. In rari casi, la fibrosi cistica può manifestarsi come un disturbo della coagulazione del sangue. I bambini piccoli sono particolarmente sensibili ai disturbi da malassorbimento di vitamina K perché solo una piccola quantità di questa vitamina attraversa la placenta, lasciando il bambino con riserve molto basse. Poiché i fattori II, VII, IX e X (fattori di coagulazione) sono vitamina K-dipendenti, bassi livelli di essa possono causare problemi. La patologia polmonare è la conseguenza dell’ostruzione delle vie aeree causata dall’accumulo di muco, dalla riduzione della clearance mucociliare e dall’infiammazione.
Polmoni
L’infiammazione e l’infezione causano lesioni e cambiamenti strutturali per i polmoni, portando ad una varietà di sintomi. Nelle fasi iniziali, tosse incessante, produzione abbondante di espettorato e ridotta capacità polmonare, sono condizioni comuni. Molti di questi sintomi si verificano quando i batteri, che normalmente abitano l’espettorato denso, crescono fuori controllo e causano polmonite. Nelle fasi successive, i cambiamenti nella struttura del polmone, come le patologie delle vie aeree principali (bronchiectasie), aggravano ulteriormente le difficoltà nella respirazione. Altri sintomi includono tosse con sangue (emottisi), alta pressione sanguigna nei polmoni (ipertensione polmonare), insufficienza cardiaca, difficoltà ad ottenere abbastanza ossigeno (ipossia) e insufficienza respiratoriache richiede il supporto con maschere respiratorie. Lo Staphylococcus aureus, l’Haemophilus influenzae e il Pseudomonas aeruginosa sono i tre organismi più comuni che causano infezioni polmonari nei pazienti con la fibrosi cistica. Oltre alle tipiche infezioni batteriche, le persone con la condizione, solitamente, sviluppano altri tipi di malattie polmonari. Tra queste l’aspergillosi broncopolmonare allergica, in cui la risposta del corpo al comune fungo Aspergillus fumigatus provoca un peggioramento dei problemi respiratori. Un’altra patologia è l’infezione da Mycobacterium avium complex (MAC), un gruppo di batteri legati alla tubercolosi, che possono causare danni ai polmoni e non rispondono agli antibiotici comuni. Il muco presente nei seni paranasali è ugualmente denso e può anche causare il blocco dei passaggi, con conseguente infezione. Questo può causare dolore facciale, febbre, scolo nasale, cefalea e aumentare le difficoltà respiratorie. Soggetti con fibrosi cistica possono sviluppare crescita eccessiva del tessuto nasale (Poliposi naso-sinusale) a causa di infiammazione da infezioni croniche dei seni. Polipi ricorrenti possono verificarsi in circa il 10% al 25% dei pazienti affetti da fibrosi cistica. Le complicanze cardiorespiratorie sono la causa più comune di morte (~ 80%) nei pazienti affetti dalla condizione.
Leggi anche:
- Sclerodermia: cause, sintomi e cura
- Sindrome di Sjögren: sintomi, invalidità, terapia e mortalità
- Fibromialgia: sintomi, cause, cura e tender points
Segni e sintomi gastrointestinali
Prima dello screening prenatale e neonatale, la fibrosi cistica era spesso diagnosticata quando un neonato non era in grado di espellere le feci (meconio). Il meconio può bloccare completamente l’intestino e causare gravi malattie. Questa condizione, detta ileo da meconio, si verifica nel 5-10% dei neonati con fibrosi cistica. Inoltre, la protrusione della membrana interna rettale (prolasso rettale) è più comune, e si verifica in circa il 10% dei bambini affetti dalla condizione, ed è causata da un aumento del volume fecale, dalla malnutrizione e da una maggiore pressione intra-addominale causata dalla tosse.
Pancreas
Le secrezioni anomale nel pancreas bloccano il movimento degli enzimi digestivi nel duodeno e provocano danni irreversibili al pancreas, spesso sfociando in una dolorosa infiammazione (pancreatite). Nei casi più gravi e avanzati, i dotti pancreatici appaiono atrofici.
Leggi anche:
- Pancreatite acuta: terapia, dieta, complicanze, morte
- Pancreatite cronica: sintomi, diagnosi, cura, dieta, mortalità
L’insufficienza pancreatica esocrina si verifica nella maggior parte (dall’85% al 90%) dei pazienti con fibrosi cistica. È principalmente associata con “gravi” mutazioni del gene CFTR, in cui entrambi gli alleli sono completamente non funzionali (ad es ΔF508/ΔF508). Si verifica nel 10-15% dei pazienti con una mutazione “grave” e una “media” del gene CFTR, dove c’è ancora una lieve attività dell’CFTR o dove vi sono due “medie” mutazioni. In questi casi più lievi, vi è ancora sufficiente funzione esocrina del pancreas in modo che la supplementazione di enzimi non sia necessaria.
Leggi anche: Insufficienza pancreatica: esami, cause, sintomi, cura, dieta
Le secrezioni dense inoltre possono causare problemi al fegato. La bile secreta per aiutare la digestione può bloccare i dotti biliari, causando danni epatici. Nel corso del tempo, questo può portare a cicatrici e nodularità (cirrosi). Il fegato non riesce a liberare il sangue dalle tossine e non sintetizza le proteine importanti, come quelle responsabili della coagulazione del sangue. Le patologie epatiche sono la terza causa più comune di morte correlata alla fibrosi cistica.
Leggi anche: Cirrosi epatica e fegato: sintomi, dieta, diagnosi, terapia e prevenzione
Oltre ai problemi del pancreas, le persone con fibrosi cistica lamentano bruciore di stomaco, blocco intestinale da intussuscezione e costipazione. Gli individui anziani affetti da fibrosi cistica possono sviluppare ostruzione intestinale distale a causa delle feci ispessite.
Leggi anche:
- Differenze tra ileo meccanico ed ileo paralitico: cause, sintomi e trattamenti
- Fecaloma: tappo di feci durissime, cause, sintomi e rimedi
- Defecografia: cos’è, a che serve, come ci si prepara, è dolorosa?
- Fecaloma ed ostruzione intestinale: quando chiamare il medico
- Feci dure, stitichezza e dolore defecazione: cause e cure
Malnutrizione
La mancanza di enzimi digestivi porta a difficoltà di assorbire i nutrienti, con la loro successiva escrezione nelle feci: il “malassorbimento”. Il malassorbimento porta alla malnutrizione per difetto e alla scarsa crescita. L’ipoproteinemia risultante può essere abbastanza grave da provocare edema generalizzato. Gli individui affetti da fibrosi cistica hanno anche difficoltà di assorbire le vitamine liposolubili A, D, E e K.
Leggi anche:
- Malnutrizione per difetto o eccesso: definizione, sintomi, significato
- Ipovitaminosi (carenza di vitamine): sintomi, lingua, unghie, cura
- Carenza di vitamina A (retinolo): sintomi, dieta e integratori consigliati
- Carenza di vitamina D, rachitismo, osteomalacia: cause, sintomi, terapie, dieta, integratori
- Carenza di vitamina K: sintomi, fabbisogno, dieta e integratori consigliati
- Carenza di niacina e triptofano: pellagra, cause, terapie e dieta
- Carenza e dipendenza da vitamina B6: cause, sintomi, terapie, dieta, integratori
- Carenza di biotina: cause, sintomi, fabbisogno, dieta, integratori
- Carenza di vitamina C e scorbuto: cause, sintomi, cura, dieta, integratori
- Carenza di acido pantotenico (vitamina B5): cause, sintomi, cure, dieta, integratore
- Ipoproteinemia (basso livello di proteine nel sangue): cause e conseguenze
- Iperproteinemia (alto livello di proteine nel sangue): cause e conseguenze
Diabete nella fibrosi cistica
I danni del pancreas possono portare alla perdita delle cellule insulari, causando una forma di diabete caratteristica degli affetti di fibrosi cistica. Ciò è una delle più importanti complicanze non polmonari della malattia. Il diabete è la complicanza non-polmonare più frequente nei casi di fibrosi cistica. Viene riconosciuto come una entità distinta, e presenta caratteristiche miste del tipo 1 e del tipo 2. Nonostante sia utilizzati farmaci orali contro il diabete, l’unico trattamento raccomandato consiste in iniezioni di insulina o l’uso di una pompa di insulina. A differenza del diabete classico, non vengono raccomandate restrizioni nella dieta.
Leggi anche:
- Differenze tra il diabete di tipo 1 e 2 (insulino dipendente e resistente)
- Diabete mellito: diffusione, sintomi, classificazione e diagnosi differenziale
- Diabete mellito: cause e fattori di rischio del tipo 1 e 2
- Diabete mellito: come si forma la malattia?
- Diabete mellito: conseguenze e complicanze a lungo termine
- Prevenzione del diabete mellito
Vitamina D, osteoporosi e dita ippocratiche
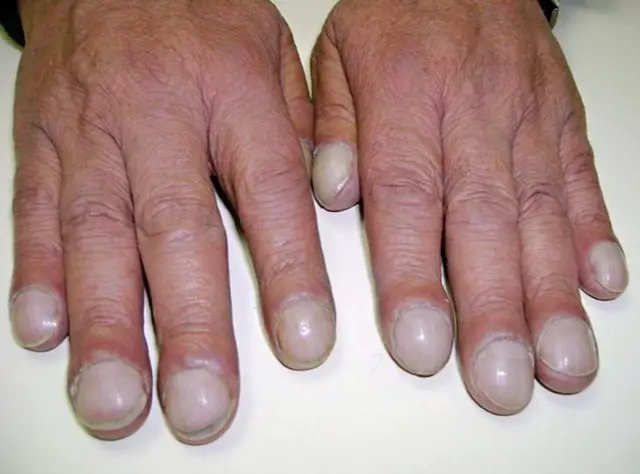
La vitamina D è coinvolta nella regolazione del calcio e del fosfato. Lo scarso assorbimento della vitamina D nella dieta, a causa del malassorbimento, può portare a osteoporosi, una condizione in cui le ossa indebolite sono più soggette a fratture. Inoltre, le persone con fibrosi cistica sviluppano spesso le cosiddette dita ippocratiche (dette anche clubbing) a causa del basso tenore di ossigeno nei loro tessuti; a tal proposito, leggi anche: Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
Infertilità
L’infertilità interessa sia gli uomini che le donne. Almeno il 97% degli uomini affetti da fibrosi cistica sono infertili, ma non sterili e possono avere figli con tecniche di procreazione assistita. La principale causa di infertilità negli uomini affetti da fibrosi cistica è l’assenza congenita dei vasi deferenti (che normalmente connettono i testicoli ai dotti eiaculatori del pene), ma potenzialmente vi possono essere anche ulteriori problemi che possono causare azoospermia, teratospermia e oligoastenospermia. Alcune donne hanno difficoltà di procreazione a causa dell’ispessimento del muco cervicale o per via della malnutrizione. Nei casi più gravi, la malnutrizione interrompe l’ovulazione e causa amenorrea. Leggi anche:
- Differenza tra infertilità e sterilità
- Astenospermia: spermiogramma, spermatozoi deboli e fattori che influenzano la loro motilità
- Ipoposia: quando lo sperma è troppo poco. Cause e terapie per aumentare la quantità di eiaculato
Diagnosi: anamnesi
La fibrosi cistica viene inizialmente riconosciuta durante l’infanzia o la fanciullezza; tuttavia, in una piccola parte di pazienti, la diagnosi viene posta in età adulta. I pazienti affetti da fibrosi cistica, frequentemente, manifestano innanzitutto infezioni polmonari ricorrenti.
I bambini con fibrosi cistica sono affetti da infezioni respiratorie più frequenti e prolungate rispetto ai bambini normali. La maggior parte dei bambini con fibrosi cistica presenta tosse cronica e sibili. Con il progredire della malattia diventano prominenti i sintomi correlati alle bronchiectasie e all’ipereattività bronchiale. Si osservano anche ippocratismo digitale e dispnea da sforzo. La febbre è per lo più di lieve entità durante l’esacerbazione delle bronchiectasie, ma può essere molto elevata durante gli episodi di polmonite. Nelle fasi avanzate della malattia le complicanze dovute al coinvolgimento polmonare comprendono l’emoftisi, che è occasionalmente massiva, lo pneumotorace, l’atelettasia, il cuore polmonare e l’insufficienza respiratoria.
Il coinvolgimento pancreatico in presenza di fibrosi cistica provoca insufficienza pancreatica esocrina. La mancanza di enzimi pancreatici provoca digestione difficoltosa e malassorbimento. L’insuffìcienza pancreatica esocrina è associata a diarrea e feci che contengono grandi quantità di grassi. Questi sintomi sono frequentemente associati a dolori addominali crampiformi, malnutrizione ed incapacità a mantenere una velocità di crescita adeguata. Altri sintomi gastrointestinali rilevabili all’anamnesi meno comuni includono tappi di meconio, intussuscezione (scivolamento di una parte dell’intestino in un’altra parte); prolasso rettale, ostruzione intestinale, ittero neonatale prolungato, cirrosi epatica, colelitiasi (calcoli biliari in colecisti); pancreatiti ricorrenti e diabete mellito.
Le anomalie della produzione di sudore si manifestano con una elevata concentrazione di sali nel sudore. Questo aumento di sali provoca un sapore salato della cute e lo sviluppo di cristalli di sale sulla cute o all’interno degli abiti, in particolare nelle scarpe e negli stivali. La perdita degli elettroliti durante i mesi estivi può provocare una intolleranza al caldo, prostrazione da caldo, deplezione degli elettroliti e disidratazione.
All’anamnesi, i sintomi correlati alla porzione superiore dell’apparato respiratorio includono le sinusiti ricorrenti e lo sviluppo di polipi nasali. Quasi tutti gli uomini e la maggior parte delle donne con fibrosi cistica sono sterili. Se una donna con fibrosi cistica diviene gravida, non è sicuro che conduca a termine la gravidanza. Il nascituro sarà affetto da fibrosi cistica o sarà portatore del gene della fibrosi cistica.
Diagnosi: esame obiettivo
L’esame obiettivo dei pazienti affetti da fibrosi cistica è quasi sempre anormale entro alcuni anni dalla diagnosi della malattia. I pazienti sono, di solito, bambini magri o giovani adulti. Se è presente distress respiratorio, saranno utilizzati i muscoli accessori della respirazione. La tosse produttiva è un reperto quasi universale. l’esame delle estremità può evidenziare ippocratismo digitale. I’esame delle vie aeree superiori rivela la presenza di polipi nasali o dolorabilità in corrispondenza dei seni paranasali. Il torace sembra avere una configurazione a botte. I polmoni presentano grossolani crepitii e sibili. Nella fase avanzata della malattia l’ipossiemia si manifesta con cianosi intorno alla bocca. L’auscultazione del cuore può evidenziare una componente polmonare del secondo tono, indice di ipertensione polmonare. La distensione delle vene giugulari del collo e l’edema pedidio sono associati allo sviluppo dell’insufficienza della sezione destra del cuore (cuore polmonare).
Diagnosi: esami di laboratorio
All’emogasanalisi, i valori emogasanalitici sono quasi normali nelle fasi iniziali della malattia a parte un aumento del gradiente alveolo-arterioso di ossigeno. L’ipossiemia in aria ambiente aumenta con il progredire della malattia, mentre l’ipercapnia e l’ipossiemia severa compaiono soltanto nelle fasi molto avanzate della malattia polmonare. Nella fibrosi cistica i reperti biochimici sierici e l’esame emocromocitometrico non mostrano, invece, alterazioni tipiche. Comunque, è possibile osservare una elevazione dei bicarbonati sierici in conseguenza di una insufficienza respiratoria cronica mentre l’elevazione dell’ematocrito può essere il riflesso dell’ipossiemia cronica. La comparsa di una broncopolmonite acuta può portare alla elevazione dei globuli bianchi con una conseguente comparsa delle forme più immature di granulociti. La concentrazione di proteine sieriche ed albumina può essere, invece, bassa in presenza di malnutrizione. La misurazione del contenuto elettrolitico del sudore ha costituito la tecnica standard per la conferma diagnostica della fibrosi cistica. Dopo aver stimolato la secrezione del sudore esso viene raccolto a tenuta stagna e dopo la raccolta di circa 0.1 ml di sudore, si effettua la misurazione del contenuto elettrolitico. In età pediatrica la diagnosi di fibrosi cistica viene posta in presenza di una concentrazione di cloro nel sudore superiore a 60 mEq/l, mentre in età adulta per poter porre tale diagnosi è necessario raggiungere una concentrazione di cloro superiore ad 80 mEq/l.
Se la risposta a questo esame è dubbia (tra 50 ed 80 mEq/l), la ripetizione di tale misurazione può risolvere il dubbio diagnostico. Sebbene la concentrazione elettrolitica nel sudore sia utile per confermare la diagnosi di fibrosi cistica, tale valutazione deve essere effettuata con meticolosa attenzione oppure i risultati possono essere fonte di confusione. I pazienti affetti da fibrosi cistica presentano tipicamente microrganismi patogeni nell’espettorato; i 3 più frequentemente riscontrati sono lo Staphylococcus aureus, l’Haemophilus influenzae e la Pseudomonas aeruginosa. Il ceppo
di P. aeruginosa ritrovato nei pazienti affetti da fibrosi cistica produce, tipicamente, mucina. Questa forma mucoide di P. aeruginosa è quasi unicamente riscontrata in pazienti con fibrosi cistica e si evidenzia quasi esclusivamente nelle vie aeree di coloro che sono affetti da forme avanzate di tale malattia. Le esacerbazioni gravi possono essere comunque causate da numerosi diversi ceppi di P. aeruginosa produttori di mucina.
Diagnosi: spirometria
I test di funzionalità polmonare sono molto utili nel valutare l’estensione del processo patologico polmonare e nel seguire la velocità di progressione. Seguendo la progressione del processo patologico si consente al clinico di aumentare la terapia nel caso in cui la funzionalità polmonare si deteriori in maniera inattesa. La spirometria mostra tipicamente l’ostruzione delle vie aeree con riduzione del volume di espirazione forzata in 1 secondo (FEV1), mentre nelle fasi avanzate della malattia è possibile osservare, invece, la perdita della capacità vitale forzata (FVC). Entrambe queste modificazioni possono, però, migliorare dopo la somministrazione di broncodilatatori. Il volume residuo aumenta precocemente nel corso della malattia e può essere validamente misurato utilizzando un pletismografo corporeo.
Diagnosi: diagnostica per immagini
La radiografia del torace mostra caratteristicamente una iperespansione, diagnosticata dall’appiattimento degli emidiaframmi e dall’incremento dello spazio aereo retrosternale. È di comune osservazione, inoltre, l’ispessimento della parete bronchiale sotto forma di linee parallele che si irradiano all’esterno a partire dall’ilo polmonare e
che vengono chiamate a “binario di treno”. Vengono inoltre evidenziate nella periferia del polmone piccole opacità rotondeggianti, le quali possono rappresentare piccoli ascessi situati distalmente alle vie aeree ostruite. Queste aree solitamente vanno incontro a detersione lasciando piccole cisti residue del processo infettivo. Altre anomalie osservabili alla radiografia del torace comprendono le atelettasie, la fibrosi, l’adenopatia ilare, la broncopolmonite e lo pneumotorace.
Diagnosi: test genetici
La scoperta del gene della fibrosi cistica ha, inoltre, reso possibile l’esecuzione di test diagnostici genetici per alcune anomalie genetiche associate alla fibrosi cistica. La valutazione del solo gene delta F508 permetterà, infatti, di identificare circa il 70% dei geni anomali oppure circa il 50% dei pazienti affetti. Poiché il test non è, comunque, in grado di identificare il 100 dei geni della fibrosi cistica, esso viene più correttamente riservato per la valutazione dei pazienti con sospetta fìbrosi cistica che mostrano risultati dubbi al test del sudore oppure di soggetti che necessitano di consulenza genetica in quanto ad elevato rischio per il concepimento di figli con fibrosi cistica.
Per approfondire:
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Fibrosi cistica: storia, organi coinvolti, cause, trasmissione, anatomia patologica
- Fibrosi cistica: terapia farmacologica e chirurgica, prognosi, mortalità
Leggi anche:
- Insufficienza respiratoria acuta e cronica: cause e conseguenze
- Insufficienza polmonare lieve, severa, acuta: sintomi e cura
- Differenza tra insufficienza respiratoria di tipo 1 e 2
- Saturazione dell’ossigeno: valori normali e patologici in anziani e bambini
- I migliori saturimetri professionali per uso ospedaliero e casalingo
- Emogasanalisi arterioso: procedura, interpretazione, è dolorosa?
- Ipossiemia: significato, valori, sintomi, conseguenze, rischi, cure
- Ipercapnia: valori, terapia, conseguenze e trattamento
- Ipossia: valori, conseguenze, sintomi, cure
- Anossia: definizione, cause, sintomi, sinonimo, cure
- Ipocapnia: significato, cause, valori, alcalosi respiratoria
- Differenza tra ipossiemia e ipercapnia
- Differenza tra ipossiemia, ipossia, anossiemia ed anossia
- Alterazioni dell’equilibrio acido-base: acidosi ed alcalosi respiratorie e metabolica
- Differenza tra acidosi ed alcalosi, metabolica e respiratoria
- Sindrome da distress respiratorio (ARDS): definizione e linee guida
- Edema polmonare acuto, cardiogeno, cause, sintomi e terapie
- Radiografia del torace: come si fa, indicazioni, bisogna spogliarsi, costo
- Broncopneumopatia cronica ostruttiva (BPCO): sintomi, diagnosi e cura
- Broncoscopia polmonare con biopsia: a cosa serve, fa male, è pericolosa?
- Tumore al polmone: aspettativa di vita, sopravvivenza, fase terminale
- Tumore pleurico e mesotelioma: sopravvivenza e aspettativa di vita
- Cavo, liquido e versamento pleurico: fisiologia e patologia
- Differenza tra PM10 e PM2,5 e rispettivi effetti sulla salute
- Da oggi l’inquinamento dell’aria è ufficialmente un cancerogeno
- Polvere toracica: le case dei fumatori sono inquinate come Pechino
- Consigli per la prevenzione delle sindromi asmatiche
- Attività fisica e asma bronchiale: sport consigliati e sconsigliati
- Asma bronchiale estrinseca, intrinseca, occupazionale, stabile: cause, sintomi, cure
- Asma bronchiale in bambini e adulti: cause, sintomi e cura
- Test di provocazione bronchiale con metacolina: esecuzione, preparazione, rischi
- Iper-reattività bronchiale: significato, sintomi, diagnosi e cure
- Asma occupazionale: cause, sintomi, diagnosi e terapie
- Malattie respiratorie occupazionali o professionali: effetti, elenco, tipi
- Visita allergologica: svolgimento, esami, preparazione, durata, costo
- Differenza tra allergia, pseudoallergia e intolleranza alimentare
- Consigli per la prevenzione delle allergie inalatorie da pollini
- Broncopolmonite: sintomi iniziali, contagio, prognosi, morte
- Polmonite ab ingestis: cause, tempi di guarigione, morte, sopravvivenza
- Tracheite virale, batterica, allergica: quanto dura e come si cura
- Laringite acuta e cronica: antibiotico, catarrale, cure, rimedi naturali
- Faringite virale e batterica: rimedi, quanto dura, è contagiosa?
- Bronchite: sintomi, durata, cura, rimedi naturali, è contagiosa?
- Differenza tra bronchite acuta e cronica
- Differenza tra bronchite e polmonite
- Mal di gola forte: rimedi naturali e farmaci per farlo passare
- Cordite in medicina: significato, da reflusso, sintomi, cura
- Laringoscopia diretta e indiretta: anestesia, costo, è dolorosa?
- Differenza tra laringoscopia diretta, indiretta, rigida, flessibile
- Intossicazione da monossido di carbonio: sintomi, danni permanenti, morte
- Atelettasia: significato, polmonare, cause, sintomi, cura, riabilitazione
- Laringospasmo: virale, da ansia, da stress, significato
- Broncospasmo in bambini e anziani: rimedi, quanto dura, paradosso
- Differenza tra laringospasmo e broncospasmo
- Morte per soffocamento: segni, sintomi, fasi e tempi
- Cosa si prova a morire annegati, dissanguati, decapitati…
- Differenze tra faringite, laringite e tracheite: vari tipi di mal di gola
- Laringite ipoglottica (pseudocroup): cause, sintomi, cosa fare
- Croup nel bambino: significato, cause, sintomi, terapia, mortalità
- Differenza tra croup e pseudocroup
- Bronchiolite in neonati e bambini: sintomi, cause, è pericolosa?
- Bronchiolite nei bimbi: mortalità, pericoli, complicazioni e durata
- Tampone faringeo: preparazione, digiuno, positivo o negativo
- Differenza tra tonsillite, faringite, laringite, tracheite, adenoidite
- Differenza tra mal di gola e tonsillite
- Differenza tra mal di gola virale e batterico
- Abbassamento della voce: cause, rimedi, quando andare dal medico
- Polipi, noduli e granulomi delle corde vocali: cause, sintomi e cure
- Dove si trovano le corde vocali: anatomia e funzioni in sintesi
- Sensazione di nodo alla gola in medicina: cause, sintomi, diagnosi, cure
- Perché viene la tosse e come faccio a farla passare?
- Perché si starnutisce? Cosa fare se si continua a starnutire?
- Differenza tra tosse secca, grassa, cronica e con catarro
- Perché ci viene la febbre e perché non dobbiamo aver paura di lei
- Differenza tra raffreddore e influenza: sintomi comuni e diversi
- Come e quando abbassare la febbre? Farmaci e rimedi casalinghi
- Tonsille gonfie, infiammate e con placche rimedi in adulti e bambini
- Tonsillectomia: cosa mangiare, convalescenza, dolore, in adulti e bambini
- Tonsille palatine, linguali, faringee e tubariche: funzioni e patologia
- Differenza tra tonsille e adenoidi
- Adenoidi: ingrossate, operazione, convalescenza, ricrescono?
- Adenoidite acuta e cronica in bambini ed adulti: cause, sintomi, cure
- Differenze tra Paracetamolo, Tachipirina, Ibuprofene, Aspirina, Efferalgan e Co-Efferalgan
- Asfissia: sintomi, cure ed in quanto tempo si muore
- Respirazione artificiale bocca a bocca: quando farla e come farla
- Primo soccorso e BLS (Basic Life Support): cos’è e come si fa
- Cianosi in volto, mani o labbra: significato, cause, rischi, cure
- Differenza tra Mucosolvan e Fluimucil
- Spirometria diretta ed indiretta: come si esegue ed a cosa serve
- Sì al bagno dopo mangiato, ecco le vere 8 cause più frequenti di morte in acqua
- Croup nel bambino: significato, cause, sintomi, terapia, mortalità
- Differenza tra asfissia e soffocamento
- Differenza tra soffocamento e strangolamento
- Massaggio cardiaco: quando farlo e come farlo [LINEE GUIDA]
- Morte in culla (SIDS): prevenzione, cause, sintomi e percentuale dei casi
- Malattia da decompressione: terapia e fisiopatologia
- Embolia gassosa arteriosa da immersione: sintomi e cure
- Cosa succede al cibo nello stomaco dopo averlo ingerito?
- Apnea ostruttiva del sonno: cause, rischi, trattamenti e prevenzione
- Perché si russa e quali sono i rimedi per smettere di russare? I pericoli dell’apnea ostruttiva del sonno
- Crisi respiratoria acuta e rischio di morte: cosa fare?
- Pallore in viso: significato, sinonimo, cause, ansia, cosa mangiare
- Le 7 fasi della deglutizione (volontarie ed involontarie)
- Differenza tra disfagia di tipo ostruttivo e di tipo motorio
- Differenza tra disfagia ai liquidi e ai solidi
- Insufficienza respiratoria acuta e cronica: cause e conseguenze
- Intubazione: rischi, anestesia, rianimazione, dolore alla gola
- Tracheotomia possibilità di parlare, durata, conseguenze, quando si fa
- Tracheostomia: complicanze, parlare, percutanea, cibo e gestione
- Differenza tra tracheostomia percutanea e tradizionale (a cielo aperto)
- Differenza tra tracheotomia e tracheostomia
- Cricotiroidotomia: urgenza, complicanze, procedura ed indicazioni
- Differenza tra tracheotomia e cricotiroidotomia
- Come si misura la frequenza respiratoria?
- Frequenza respiratoria normale, alta, bassa, a riposo e sotto sforzo
- Differenze tra respiro normale e patologico
- Iperventilazione: significato, sintomi, alcalosi e conseguenze
- Sindrome da iperventilazione cronica: cause, sintomi, diagnosi, cure
- Dispnea ansiosa, notturna e cardiaca: sintomi, diagnosi e cura
- Differenza tra dispnea, apnea e tachipnea
- Enfisema polmonare: sintomi, tipi, cause, diagnosi e terapia
- I migliori elettrocardiografi ed Holter portatili
- I migliori sfigmomanometri e stetoscopi per misurare la pressione arteriosa
- I migliori glucometri di ultima generazione per misurare la glicemia
- I migliori misuratori di colesterolo LDL HDL e trigliceridi, affidabili e precisi
- Fenomeno di Raynaud: cause, sintomi e trattamento
- Cos’è l’edema, come e perché si forma?
- Pneumotorace spontaneo primario, secondario ed iperteso: cause, sintomi, terapie
- Dipendenza da nicotina: come smettere di fumare sigarette in 20 passi
- Stop alle sigarette: i migliori farmaci per smettere di fumare
- Champix compresse per smettere di fumare: foglio illustrativo, prezzo
- Bupropione per smettere di fumare: meccanismo d’azione, posologia
- Gomme e cerotti: i sostituti con nicotina per smettere di fumare
- Gomme, farmaci, cerotti… Qual è il metodo migliore per smettere di fumare?
- Smettere di fumare: meglio diminuire le sigarette o interrompere di colpo?
- Vareniclina per emettere di fumare: meccanismo d’azione, efficacia
- Champix: funziona davvero per smettere di fumare?
- Respiro patologico: le alterazioni del ritmo respiratorio normale
- Respiro di Biot ed apnee: caratteristiche e cause patologiche e non patologiche
- Respiro di Cheyne-Stokes: caratteristiche e cause patologiche e non patologiche
- Respiro di Falstaff: caratteristiche e cause
- Respiro di Kussmaul: caratteristiche e cause
- Idrotorace: cause, patologie, sintomi, diagnosi e cure
- Embolia polmonare: massiva, diagnosi, da tumore, terapia
- Polmoniti nosocomiali: cause, terapie e linee guida ATS
- Polmonite interstiziale, atipica, senza febbre: sintomi e cure in bimbi ed adulti
- Tumore al polmone operabile ed inoperabile: stadiazione
- Massaggio cardiaco: quante compressioni al minuto?
- Toracentesi: procedura, complicanze, rischi, è dolorosa?
- Parametri della spirometria: capacità, volumi, rapporti e flussi
- Empiema pleurico: guarigione, saccato, complicanze, RX, cura
- Differenza tra dispnea ed affanno
- Differenza tra polipnea e tachipnea
- Tipologie di respirazione nello yoga
- Apparato respiratorio: anatomia in sintesi, struttura e funzioni
- Trachea: anatomia e funzioni in sintesi
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Differenza tra inspirazione e espirazione: l’atto respiratorio
- Differenza tra ventilazione polmonare e alveolare: spazio morto anatomico e fisiologico
- I polmoni fanno male: i sintomi di una malattia polmonare
- Dove si trovano i polmoni ed a che servono?
- Esaminare il polmone con le prove di funzionalità respiratoria
- Ipertensione polmonare: lieve, severa, terapia, aspettativa di vita
- Si può vivere senza uno o entrambi i polmoni?
- Si può vivere senza respirare aria? Quanto può durare una apnea?
- Si può vivere di sola aria senza mangiare né bere? Per quanto tempo?
- Ossigenoterapia: uso, controindicazioni, domiciliare, con maschera
- Ventilazione meccanica o artificiale: tipi ed indicazioni
- Differenza tra ventilazione meccanica e ossigenoterapia
- Pallone AMBU: a cosa serve, componenti, scadenza, percentuale di ossigeno
- Pallone va e vieni: cos’è, come funziona, a che serve
- Differenza tra pallone AMBU e va e vieni: vantaggi e svantaggi
- Cannula nasale per ossigenoterapia: cos’è, come è fatta, quando si usa
- Sondino nasale per ossigenoterapia: cos’è, come è fatta, quando si usa
- Differenza tra NIV, CPAP e BIBAP
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
