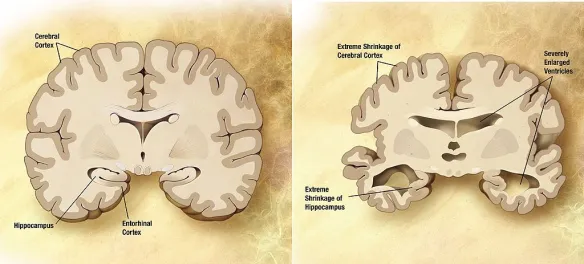
 La malattia di Alzheimer, precedentemente denominata morbo di Alzheimer o semplicemente Alzheimer, è la forma più comune di demenza degenerativa progressivamente invalidante con esordio prevalentemente in età presenile (oltre i 65 anni, ma può manifestarsi anche in epoca precedente). Si stima che circa il 60-70% dei casi di demenza sia dovuta a tale condizione. Il sintomo precoce più frequente è la difficoltà nel ricordare eventi recenti. Con l’avanzare dell’età possiamo avere sintomi come: afasia, disorientamento, cambiamenti repentini di umore, depressione, incapacità di prendersi cura di sé, problemi nel comportamento. Ciò porta il soggetto inevitabilmente a isolarsi nei confronti della società e della famiglia. A poco a poco, le capacità mentali basilari vengono perse. Anche se la velocità di progressione può variare, l’aspettativa media di vita dopo la diagnosi è dai tre ai nove anni. La patologia è stata descritta per la prima volta nel 1906, dallo psichiatra e neuropatologo tedesco Alois Alzheimer. Nel 2006 vi erano 26,6 milioni di malati in tutto il mondo e si stima che ne sarà affetta 1 persona su 85 a livello mondiale entro il 2050.
La malattia di Alzheimer, precedentemente denominata morbo di Alzheimer o semplicemente Alzheimer, è la forma più comune di demenza degenerativa progressivamente invalidante con esordio prevalentemente in età presenile (oltre i 65 anni, ma può manifestarsi anche in epoca precedente). Si stima che circa il 60-70% dei casi di demenza sia dovuta a tale condizione. Il sintomo precoce più frequente è la difficoltà nel ricordare eventi recenti. Con l’avanzare dell’età possiamo avere sintomi come: afasia, disorientamento, cambiamenti repentini di umore, depressione, incapacità di prendersi cura di sé, problemi nel comportamento. Ciò porta il soggetto inevitabilmente a isolarsi nei confronti della società e della famiglia. A poco a poco, le capacità mentali basilari vengono perse. Anche se la velocità di progressione può variare, l’aspettativa media di vita dopo la diagnosi è dai tre ai nove anni. La patologia è stata descritta per la prima volta nel 1906, dallo psichiatra e neuropatologo tedesco Alois Alzheimer. Nel 2006 vi erano 26,6 milioni di malati in tutto il mondo e si stima che ne sarà affetta 1 persona su 85 a livello mondiale entro il 2050.
Leggi anche: Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
La causa e la progressione della malattia di Alzheimer non sono ancora ben compresi. La ricerca indica che la malattia è strettamente associata a placche amiloidi e ammassi neurofibrillari riscontrati nel cervello, ma non è nota la causa prima di tale degenerazione. Attualmente i trattamenti terapeutici utilizzati offrono piccoli benefici sintomatici e possono parzialmente rallentare il decorso della patologia; anche se sono stati condotti oltre 500 studi clinici per l’identificazione di un possibile trattamento per l’Alzheimer, non sono ancora stati identificati trattamenti che ne arrestino o invertano il decorso. Circa il 70% del rischio si ritiene sia genetico con molti geni solitamente coinvolti. Altri fattori di rischio includono: traumi, depressione o ipertensione. Il processo della malattia è associata a placche amiloidi che si formano nel SNC.
Una diagnosi probabile è basata sulla progressione della malattia, test cognitivi con imaging medico e gli esami del sangue per escludere altre possibili cause. I sintomi iniziali sono spesso scambiati per normale invecchiamento. È necessaria la biopsia del tessuto cerebrale per una diagnosi definitiva. L’esercizio mentale e fisico possono diminuire il rischio di AD. Non esistono farmaci o integratori che scientificamente possano diminuire il rischio di AD.
A livello preventivo, sono state proposte diverse modificazioni degli stili di vita personali come potenziali fattori protettivi nei confronti della patologia, ma non vi sono adeguate prove di una correlazione certa tra queste raccomandazioni e la riduzione effettiva della degenerazione. Stimolazione mentale, esercizio fisico e una dieta equilibrata sono state proposte sia come modalità di possibile prevenzione, sia come modalità complementari di gestione della malattia.
La sua ampia e crescente diffusione nella popolazione, la limitata e, comunque, non risolutiva efficacia delle terapie disponibili e le enormi risorse necessarie per la sua gestione (sociali, emotive, organizzative ed economiche), che ricadono in gran parte sui familiari dei malati, la rendono una delle patologie a più grave impatto sociale del mondo.
Anche se il decorso clinico della malattia di Alzheimer è in parte specifico per ogni individuo, la patologia causa diversi sintomi comuni alla maggior parte dei pazienti. I primi sintomi osservabili sono spesso erroneamente considerati problematiche “legate all’età”, o manifestazioni di stress. Nelle prime fasi, il sintomo più comune è l’incapacità di acquisire nuovi ricordi e la difficoltà nel ricordare eventi osservati recentemente. Quando si ipotizza la presenza di una possibile malattia di Alzheimer, la diagnosi viene di solito confermata tramite specifiche valutazioni comportamentali e test cognitivi, spesso seguiti dall’imaging a risonanza magnetica.
Con l’avanzare della malattia, il quadro clinico può prevedere confusione, irritabilità e aggressività, sbalzi di umore, difficoltà nel linguaggio, perdita della memoria a breve e lungo termine e progressive disfunzioni sensoriali.
Poiché per la malattia di Alzheimer non sono attualmente disponibili terapie risolutive e il suo decorso è progressivo, la gestione dei bisogni dei pazienti diviene essenziale. Spesso è il coniuge o un parente stretto (caregiver), a prendersi in carico il malato, compito che comporta notevoli difficoltà e oneri. Chi si occupa del paziente può sperimentare pesanti carichi personali che possono coinvolgere aspetti sociali, psicologici, fisici ed economici.
Leggi anche: Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
La malattia di Alzheimer è definibile come un processo degenerativo che pregiudica progressivamente le cellule cerebrali, rendendo a poco a poco l’individuo che ne è affetto incapace di una vita normale e provocandone alla fine la morte. In Italia ne soffrono circa 492 000 persone e 26,6 milioni nel mondo secondo uno studio della Johns Hopkins Bloomberg School of Public Health di Baltimora, Stati Uniti, con una netta prevalenza di donne (presumibilmente per via della maggior vita media delle donne rispetto agli uomini).
Definita anche “demenza di Alzheimer”, viene appunto catalogata tra le demenze, essendo un deterioramento cognitivo cronico progressivo. Tra tutte le demenze quella di Alzheimer è la più comune, rappresentando, a seconda della casistica, l’80-85% di tutti i casi di demenza.
A livello epidemiologico, tranne che in rare forme genetiche familiari “early-onset” (cioè con esordio giovanile), il fattore maggiormente correlato all’incidenza della patologia è l’età. Molto rara sotto i 65 anni, la sua incidenza aumenta progressivamente con l’aumentare dell’età, per raggiungere una diffusione significativa nella popolazione oltre gli 85 anni.
Da rilevazioni europee, nella popolazione generale l’incidenza (cioè il numero di nuovi casi all’anno) è di 2,5 casi ogni 1 000 persone per la fascia di età tra i 65 e i 69 anni; sale a 9 casi su 1 000 persone tra i 75 e i 79 anni, e a 40,2 casi su 1 000 persone tra gli 85 e gli 89 anni.
Fasi e sintomi delle varie fasi
Il decorso della malattia è diviso in quattro fasi, con un modello progressivo deterioramento cognitivo e funzionale; per approfondire leggi: Malattia di Alzheimer: sintomi delle fasi iniziali e tardive
Cause e fattori di rischio
La causa per la maggior parte dei casi di Alzheimer è ancora in gran parte sconosciuta, ad eccezione che per casi dall’1% al 5% in cui sono state individuate le differenze genetiche esistenti. Diverse ipotesi cercano di spiegare la causa della malattia; per approfondire leggi: Malattia di Alzheimer: le cause della malattia
Decorso
Il decorso della malattia può essere diverso, nei tempi e nelle modalità sintomatologiche, per ogni singolo paziente; esistono comunque una serie di sintomi comuni, che si trovano frequentemente associati nelle varie fasi con cui, clinicamente, si suddivide per convenzione il decorso della malattia. A una prima fase lieve, fa seguito la fase intermedia, e quindi la fase avanzata/severa; il tempo di permanenza in ciascuna di queste fasi è variabile da soggetto a soggetto, e può in certi casi durare anche diversi anni.
La malattia viene spesso anticipata dal cosiddetto mild cognitive impairment (MCI), un leggero calo di prestazioni in diverse funzioni cognitive in particolare legate alla memoria, all’orientamento o alle capacità verbali. Tale calo cognitivo, che è comunque frequente nella popolazione anziana, non è necessariamente indicativo di demenza incipiente, ma può in alcuni casi essere seguito dall’avvio delle fasi iniziali dell’Alzheimer.
La malattia inizialmente si manifesta spesso come demenza caratterizzata da amnesia progressiva e altri deficit cognitivi. Il deficit di memoria è prima circoscritto a sporadici episodi nella vita quotidiana, ovvero disturbi di quella che viene chiamata on-going memory (ricordarsi cosa si è mangiato a pranzo, cosa si è fatto durante il giorno) e della memoria prospettica (che riguarda l’organizzazione del futuro prossimo, come ricordarsi di andare a un appuntamento); poi man mano il deficit aumenta e la perdita della memoria arriva a colpire anche la memoria episodica retrograda (riguardante fatti della propria vita o eventi pubblici del passato) e la memoria semantica (le conoscenze acquisite), mentre la memoria procedurale (che riguarda l’esecuzione automatica di azioni) viene relativamente risparmiata fino alle fasi intermedio-avanzate della malattia.
A partire dalle fasi lievi e intermedie possono poi manifestarsi crescenti difficoltà di produzione del linguaggio, con incapacità nella definizione di nomi di persone od oggetti, e frustranti tentativi di “trovare le parole”, seguiti poi nelle fasi più avanzate da disorganizzazione nella produzione di frasi e uso sovente scorretto del linguaggio (confusione sui significati delle parole, ecc.). Sempre nelle fasi lievi-intermedie, la pianificazione e gestione di compiti complessi (gestione di documenti, attività lavorative di concetto, gestione del denaro, guida dell’automobile, cucinare, ecc.) cominciano a diventare progressivamente più impegnative e difficili, fino a richiedere assistenza continuativa o divenire impossibili.
Nelle fasi intermedie e avanzate, inoltre, possono manifestarsi problematiche comportamentali (vagabondaggio, coazione a ripetere movimenti o azioni, reazioni comportamentali incoerenti) o psichiatriche (confusione, ansia, depressione e, occasionalmente, deliri e allucinazioni). Il disorientamento nello spazio, nel tempo o nella persona (ovvero la mancata o confusa consapevolezza di dove si è situati nel tempo, nei luoghi e/o nelle identità personali, proprie o di altri – comprese le difficoltà di riconoscimento degli altri significativi) è sintomo frequente a partire dalle fasi intermedie-avanzate. In tali fasi si aggiungono difficoltà progressive anche nella cura della persona (lavarsi, vestirsi, assumere farmaci, ecc.).
Ai deficit cognitivi e comportamentali, nelle fasi più avanzate si aggiungono infine complicanze mediche internistiche, che portano a una compromissione progressiva della salute. Una persona colpita dalla malattia può vivere anche una decina di anni dopo la diagnosi clinica di malattia conclamata.
Come sottolineato, col progredire della malattia le persone non solo presentano deficit di memoria, ma risultano deficitarie nelle funzioni strumentali mediate dalla corteccia associativa, e possono pertanto presentare afasia e aprassia, fino a presentare disturbi neurologici e poi internistici; pertanto i pazienti, nelle fasi intermedie e avanzate, necessitano di continua assistenza personale (solitamente erogata da familiari e badanti, i cosiddetti caregiver, che sono a loro volta sottoposti ai forti stress tipici di chi assiste i malati di Alzheimer).
Leggi anche: Sclerosi multipla: cause, sintomi, diagnosi e prognosi
Diagnosi
La malattia di Alzheimer è di solito diagnosticata clinicamente dalla storia del paziente, da osservazioni cliniche, dalla presenza di particolari caratteristiche neurologiche e neuropsicologiche e per l’assenza di condizioni alternative.
Sistemi avanzati di imaging biomedico, come la tomografia computerizzata (TC), la risonanza magnetica (MRI), la tomografia a emissione di fotone singolo (SPECT) o la tomografia ad emissione di positroni (PET) possono essere utilizzate per aiutare a escludere altre patologie cerebrali o altri tipi di demenza. Inoltre, si possono prevedere il passaggio da fasi prodromiche (decadimento cognitivo lieve) alla malattia di Alzheimer.
Gli assessment neuropsicologici e cognitivi, inclusi i test di memoria ed esecutivi, possono ulteriormente caratterizzare lo stato della malattia. Diverse organizzazioni mediche hanno creato i criteri diagnostici per facilitare e standardizzare il processo diagnostico. La diagnosi clinica viene confermata a livello patologico solo con l’analisi istologica del cervello post-mortem.
Leggi anche: Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
Criteri diagnostici
Lo statunitense National Neurological Disorders and Stroke (NINCDS) e l’Associazione dei Malati di Alzheimer ha istituito il criterio diagnostico NINCDS-ADRDA nel 1984, in seguito aggiornato nel 2007. Questo criterio richiede che la presenza di deficit cognitivi e una sospetta sindrome di demenza debbano essere confermati da test neuropsicologici per porre la diagnosi clinica di Alzheimer. Una conferma istopatologica, tra cui un esame al microscopio del tessuto cerebrale (eseguibile solo post-mortem) è necessaria per una conferma della diagnosi definitiva a posteriori.
Sono otto gli ambiti funzionali cognitivi più comunemente compromessi: memoria, linguaggio, abilità percettiva, attenzione, abilità costruttiva, orientamento, risoluzione dei problemi e capacità funzionali. Questi ambiti cognitivi sono equivalenti ai criteri della NINCDS ADRDA, come elencati nel Diagnostic and Statistical Manual of Mental Disorders (DSM) pubblicato dalla American Psychiatric Association.
Tecniche diagnostiche, screening e diagnostica per immagini
Alcuni test di screening neuropsicologico e la diagnostica per immagini possono essere utili nella diagnosi di Alzheimer; per approfondire leggi: Malattia di Alzheimer: screening e diagnosi nelle fasi iniziali della malattia
Terapia
Anche se al momento non esiste una cura efficace, sono state proposte diverse strategie terapeutiche per tentare di influenzare clinicamente il decorso della malattia di Alzheimer; tali strategie puntano a modulare farmacologicamente alcuni dei meccanismi patologici che ne stanno alla base. È inoltre opportuno integrare interventi psicosociali, cognitivi e comportamentali. Per approfondire i farmaci usati nella malattia di Alzheimer: Malattia di Alzheimer: cura farmacologica
Intervento psicosociale e cognitivo
Nel paziente con malattia di Alzheimer, oltre al trattamento farmacologico, esistono interventi comportamentali, di supporto psicosociale e di training cognitivo che possono aiutare il soggetto; per approfondire leggi: Intervento psicosociale e cognitivo nel paziente con malattia di Alzheimer
Prognosi
Le fasi iniziali della malattia di Alzheimer sono difficili da diagnosticare. Una diagnosi definitiva è posta solitamente una volta che si verifica una significativa compromissione cognitiva e una percepibile riduzione di capacità di svolgere le attività della vita quotidiana, anche se la persona è ancora in grado di gestirsi autonomamente. Il deterioramento della memoria e il peggioramento dei disturbi cognitivi e non cognitivi, associati alla malattia, riducono progressivamente l’autonomia nella vita quotidiana.
L’aspettativa di vita della popolazione con la malattia si riduce, con un tempo di vita media di circa sette anni dopo la diagnosi. Meno del 3% della popolazione vive più di quattordici anni. Malattie caratteristiche significativamente associate alla ridotta sopravvivenza sono un aumento della gravità del deficit cognitivo, diminuzione del livello funzionale, diverse cadute e disturbi neurologici. Altre patologie concomitanti, come problemi cardiaci, diabete o storia di abuso di alcool sono correlate con una sopravvivenza più breve. L’aspettativa di vita è particolarmente ridotta rispetto alla popolazione sana quando la malattia di Alzheimer colpisce coloro che sono più giovani. Gli uomini hanno una prognosi di sopravvivenza meno favorevole rispetto alle donne.
La malattia è la causa di morte nel 70% dei casi. La polmonite e la disidratazione sono le cause immediate più frequenti di morte, mentre il cancro è meno frequente rispetto alla popolazione generale.
Leggi anche: Malattia di Parkinson: cause, sintomi, decorso, terapie
Prevenzione
Al momento non ci sono prove definitive per sostenere l’efficacia di una qualsiasi misura preventiva per la malattia di Alzheimer. Studi per identificarle hanno spesso prodotto risultati incoerenti. Tuttavia, studi epidemiologici hanno proposto correlazioni tra alcuni fattori modificabili (come la dieta, il rischio cardiovascolare, l’utilizzo di prodotti farmaceutici o lo svolgimento di attività intellettuali) e la probabilità per una popolazione di sviluppare la malattia. Solo ulteriori ricerche, tra cui gli studi clinici, riveleranno se questi fattori possono aiutare a prevenire o ritardare l’insorgenza della malattia di Alzheimer.
Leggi anche: Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
Quadro clinico e dieta
Sebbene i fattori di rischio cardiovascolari, come l’ipercolesterolemia, l’ipertensione, il diabete e il fumo, siano associati con un rischio maggiore di insorgenza della malattia, le statine, che sono farmaci per l’abbassamento del colesterolo, non si sono dimostrate efficaci nel prevenire o migliorare il decorso. I componenti di una dieta mediterranea, che comprendono frutta e verdura, pane, grano e altri cereali, olio d’oliva, pesce e vino rosso, possono singolarmente o tutti insieme ridurre il rischio e ritardare il decorso della malattia di Alzheimer. I loro benefici effetti cardiovascolari sono stati proposti come meccanismo di azione. Esistono prove limitate che un consumo, da lieve a moderato, di alcool, soprattutto vino rosso, sia associato a un minor rischio di Alzheimer.
Ipotesi sull’uso di vitamine non hanno trovato prove sufficienti di efficacia per raccomandare la vitamina C, E, acido folico, con o senza vitamina B12, come agenti di prevenzione o per il trattamento dell’Alzheimer. Inoltre, la vitamina E, è associata a rischi per la salute. Studi compiuti esaminando la somministrazione di acido folico (B9) e di altre vitamine B non hanno mostrato alcuna correlazione significativa con il declino cognitivo.
L’utilizzo a lungo termine di farmaci anti-infiammatori non steroidei (FANS) è associato a una ridotta probabilità di sviluppare Alzheimer. Studi post-mortem umani, studi su modelli animali, o in studi in vitro, supportano l’ipotesi che i FANS possano ridurre l’infiammazione correlata alle placche amiloidi. Tuttavia, studi riguardanti il loro uso come trattamento palliativo non sono riusciti a dimostrare risultati positivi, mentre nessun processo di prevenzione è stato realizzato. La curcumina del curry ha mostrato una certa efficacia nel prevenire i danni cerebrali, nei modelli di topo, in virtù delle sue proprietà anti-infiammatorie. La terapia ormonale sostitutiva, anche se utilizzata in passato, non è più ritenuta efficace per prevenire la demenza e in alcuni casi può anche esserne ritenuta responsabile.
Leggi anche: Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
Stile di vita
Esistono studi che mostrano correlazioni tra determinati stili di vita e l’incidenza del rischio di contrarre la patologia o con la sua progressione.
Le persone che si impegnano in attività intellettuali, come la lettura, i giochi da tavolo, i cruciverba, l’esecuzione con strumenti musicali, o che hanno una regolare interazione sociale, mostrano una riduzione del rischio di sviluppo della malattia di Alzheimer. Questo è compatibile con la teoria della riserva cognitiva, in cui si afferma che alcune esperienze di vita forniscono all’individuo una riserva cognitiva che ritarda l’insorgenza di manifestazioni di demenza. L’apprendimento di una seconda lingua, anche in tarda età, sembra ritardare la malattia di Alzheimer. La pratica di attività fisica è anch’essa un comportamento associato a un ridotto rischio di Alzheimer.
Alcuni studi hanno mostrato un aumentato rischio di sviluppare la malattia nel caso di assunzione di metalli, e, in particolare, alluminio, o in caso di esposizione a particolari solventi. La qualità di alcuni di questi studi è stata però criticata, e altri studi hanno concluso che non vi è alcuna relazione tra questi fattori ambientali e lo sviluppo di Alzheimer.
Mentre alcuni studi suggeriscono che l’esposizione a campi elettromagnetici a bassa frequenza può aumentare il rischio di sviluppare la malattia di Alzheimer, i revisori hanno rilevato che sono necessari ulteriori indagini epidemiologiche e di laboratorio per poter avvalorare tale ipotesi. Il fumo è un importante fattore di rischio per l’Alzheimer.
Alcuni studi compiuti presso il National Institute on Ageing di Baltimora ipotizzano che il digiuno a intervalli regolari (1 o 2 giorni a settimana) potrebbe avere un ruolo palliativo alle forme più gravi della malattia.
Leggi anche: Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
Costi economico-sociali
La crescente incidenza di questa patologia nella popolazione generale in tutto il mondo è accompagnata da una crescita equivalente del suo enorme costo economico e sociale: allo Stato, secondo Lancet, il costo economico per la cura dei pazienti affetti da demenza a livello mondiale è di circa 600 miliardi di dollari all’anno, con un trend di crescita che lo porterà nel 2030 ad aumentare dell’85% (e con un carico crescente anche per i Paesi in via di sviluppo), facendolo divenire uno degli oneri con maggior impatto economico per i sistemi sanitari nazionali e le comunità sociali dell’intero pianeta.
Nonostante questo, la ricerca scientifica e clinica sulla demenza è ancora gravemente sottofinanziata: in Inghilterra, ad esempio, si calcola che il costo economico complessivo della cura dei pazienti affetti da demenza superi quello per i tumori e per le malattie cardiovascolari messe insieme, ma la ricerca sulle demenze riceve solo un dodicesimo dei finanziamenti di quella per i tumori.
Leggi anche:
- Demenza senile: cause, sintomi, decorso e cure
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Differenza tra malattia di Alzheimer, demenza senile, vascolare e reversibile
- Differenza tra malattia di Alzheimer e malattia di Parkinson: sintomi comuni e diversi
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Condividi questo articolo:

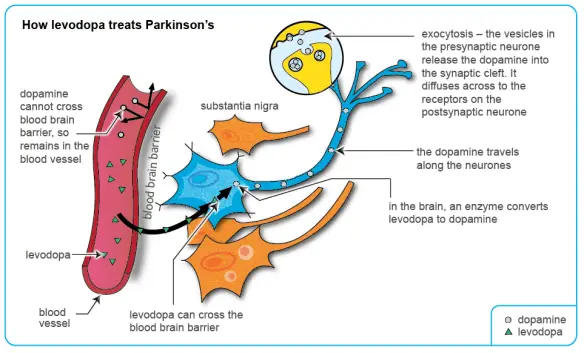 Attualmente non esiste una terapia definitiva per il morbo di Parkinson, ma il trattamento farmacologico, la chirurgia e la gestione multidisciplinare sono in grado di fornire un parziale sollievo ai sintomi. I farmaci principalmente utilizzati nel trattamento di sintomi motori sono la levodopa (di solito in combinazione con un inibitore della dopa-decarbossilasi e un inibitore delle COMT), gli agonisti della dopamina e gli inibitori MAO-B (Inibitore della monoamino ossidasi). La fase della malattia determina quale famiglia di farmaci sia più utile. Due fasi sono di solito distinte:
Attualmente non esiste una terapia definitiva per il morbo di Parkinson, ma il trattamento farmacologico, la chirurgia e la gestione multidisciplinare sono in grado di fornire un parziale sollievo ai sintomi. I farmaci principalmente utilizzati nel trattamento di sintomi motori sono la levodopa (di solito in combinazione con un inibitore della dopa-decarbossilasi e un inibitore delle COMT), gli agonisti della dopamina e gli inibitori MAO-B (Inibitore della monoamino ossidasi). La fase della malattia determina quale famiglia di farmaci sia più utile. Due fasi sono di solito distinte: La malattia di Parkinson (in passato denominata “morbo di Parkinson“; anche chiamata Parkinson, parkinsonismo idiopatico o parkinsonismo primario; in inglese denominata “Parkinson disease” o “primary parkinsonism” o “hypokinetic rigid syndrome” o “paralysis agitans” o “shaking palsy”) è una malattia neurodegenerativa extrapiramidale caratterizzata da rigidità muscolare che si manifesta con resistenza ai movimenti passivi; tremore che insorge durante lo stato di riposo e che può aumentare in caso di stato di ansia; bradicinesia che provoca difficoltà a iniziare e terminare i movimenti.
La malattia di Parkinson (in passato denominata “morbo di Parkinson“; anche chiamata Parkinson, parkinsonismo idiopatico o parkinsonismo primario; in inglese denominata “Parkinson disease” o “primary parkinsonism” o “hypokinetic rigid syndrome” o “paralysis agitans” o “shaking palsy”) è una malattia neurodegenerativa extrapiramidale caratterizzata da rigidità muscolare che si manifesta con resistenza ai movimenti passivi; tremore che insorge durante lo stato di riposo e che può aumentare in caso di stato di ansia; bradicinesia che provoca difficoltà a iniziare e terminare i movimenti.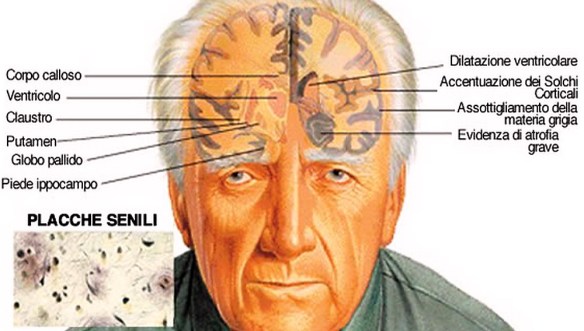 Con “demenza senile” si indica una patologia neurodegenerativa dell’encefalo, tipica dell’età avanzata e caratterizzata da una riduzione graduale ed irreversibile delle facoltà cognitive di una persona. La demenza senile rientra nella più ampia categoria delle demenze le quali sono patologie neurodegenerative dell’encefalo che possono colpire persone anziane ma non solo, e che determinano un progressivo declino delle facoltà cognitive di un individuo. E’ importante ricordare che non tutte le demenze sono irreversibili, sono reversibili ad esempio alcune forme di demenza da idrocefalo normoteso o da cause metaboliche. Un tempo “demenza senile” era sinonimo di malattia di Alzheimer. Successivamente, con la scoperta di altre forme di demenza tipiche dell’età avanzata, e con l’individuazione di una forma di Alzheimer giovanile, la dicitura demenza senile ha assunto un significato leggermente diverso, riportato nella definizione iniziale. Pur non essendo la demenza una causa diretta di morte, nella maggior parte dei casi sono i disturbi della deglutizione ad essa associata ad essere fatali per l’anziano. Tali disturbi, infatti, concorrono allo sviluppo di polmoniti da inalazione (o da aspirazione).
Con “demenza senile” si indica una patologia neurodegenerativa dell’encefalo, tipica dell’età avanzata e caratterizzata da una riduzione graduale ed irreversibile delle facoltà cognitive di una persona. La demenza senile rientra nella più ampia categoria delle demenze le quali sono patologie neurodegenerative dell’encefalo che possono colpire persone anziane ma non solo, e che determinano un progressivo declino delle facoltà cognitive di un individuo. E’ importante ricordare che non tutte le demenze sono irreversibili, sono reversibili ad esempio alcune forme di demenza da idrocefalo normoteso o da cause metaboliche. Un tempo “demenza senile” era sinonimo di malattia di Alzheimer. Successivamente, con la scoperta di altre forme di demenza tipiche dell’età avanzata, e con l’individuazione di una forma di Alzheimer giovanile, la dicitura demenza senile ha assunto un significato leggermente diverso, riportato nella definizione iniziale. Pur non essendo la demenza una causa diretta di morte, nella maggior parte dei casi sono i disturbi della deglutizione ad essa associata ad essere fatali per l’anziano. Tali disturbi, infatti, concorrono allo sviluppo di polmoniti da inalazione (o da aspirazione).
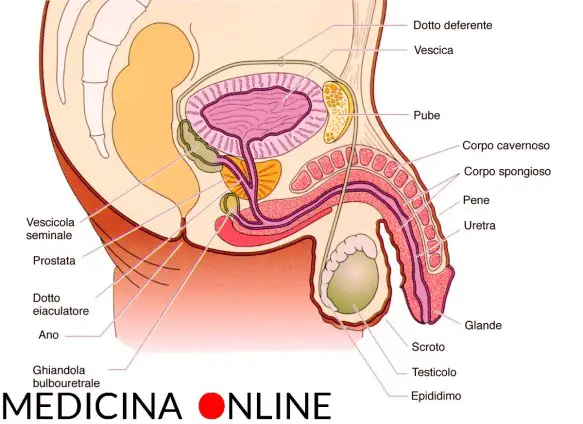
 Con testicolo ritenuto o criptorchidismo (dal greco “cripto”, nascosto) ci si riferisce ad una malformazione urologica, caratterizzata dalla mancata discesa del testicolo nel sacco scrotale. Il criptorchidismo può associarsi ad altre anomalie del tratto genito-urinario (es: ipospadia) e nel 70% dei casi riguarda il testicolo destro. Il testicolo anomalo si può trovare in un punto qualsiasi del tragitto che normalmente compie durante la vita fetale dal polo inferiore del rene allo scroto attraversando il canale inguinale. L’interruzione di questo cammino fisiologico porta ad avere il testicolo in una posizione diversa da quella normale che è quella costituita dallo scroto.
Con testicolo ritenuto o criptorchidismo (dal greco “cripto”, nascosto) ci si riferisce ad una malformazione urologica, caratterizzata dalla mancata discesa del testicolo nel sacco scrotale. Il criptorchidismo può associarsi ad altre anomalie del tratto genito-urinario (es: ipospadia) e nel 70% dei casi riguarda il testicolo destro. Il testicolo anomalo si può trovare in un punto qualsiasi del tragitto che normalmente compie durante la vita fetale dal polo inferiore del rene allo scroto attraversando il canale inguinale. L’interruzione di questo cammino fisiologico porta ad avere il testicolo in una posizione diversa da quella normale che è quella costituita dallo scroto.
 Prima di iniziare la lettura dell’articolo, vi consiglio di leggere:
Prima di iniziare la lettura dell’articolo, vi consiglio di leggere: