 I disturbi da immunodeficienza alterano la capacità del sistema immunitario di difendere l’organismo dall’invasione di cellule estranee o patologiche (come batteri, virus, funghi e cellule tumorali). Di conseguenza, si possono manifestare infezioni batteriche, virali o micotiche o linfomi oppure altri tumori normalmente poco frequenti. Un altro problema è rappresentato dal fatto che il 25% dei soggetti affetti da immunodeficienza presenta anche una malattia autoimmune (come la trombocitopenia immune). In una malattia autoimmune, il sistema immunitario attacca i tessuti stessi del corpo ( Disturbi autoimmuni). Talvolta la malattia autoimmune si sviluppa prima che l’immunodeficienza causi altri sintomi.
I disturbi da immunodeficienza alterano la capacità del sistema immunitario di difendere l’organismo dall’invasione di cellule estranee o patologiche (come batteri, virus, funghi e cellule tumorali). Di conseguenza, si possono manifestare infezioni batteriche, virali o micotiche o linfomi oppure altri tumori normalmente poco frequenti. Un altro problema è rappresentato dal fatto che il 25% dei soggetti affetti da immunodeficienza presenta anche una malattia autoimmune (come la trombocitopenia immune). In una malattia autoimmune, il sistema immunitario attacca i tessuti stessi del corpo ( Disturbi autoimmuni). Talvolta la malattia autoimmune si sviluppa prima che l’immunodeficienza causi altri sintomi.
Esistono due tipi di disturbi da immunodeficienza:
Primario: questi disturbi sono generalmente presenti alla nascita ed ereditari. Si manifestano generalmente durante la prima o la seconda infanzia. Esistono oltre 100 disturbi da immunodeficienza primari e sono tutti relativamente rari.
Secondari: questi disturbi si sviluppano generalmente più avanti nel corso della vita e spesso derivano dall’uso di certi farmaci o da un’altra malattia, come il diabete o l’infezione da virus dell’immunodeficienza umana (HIV). Sono più diffusi rispetto a quelli primari.
Alcuni disturbi da immunodeficienza abbreviano l’aspettativa di vita, mentre altri persistono per tutta la vita, ma non influiscono sulla sopravvivenza e pochi si risolvono con o senza trattamento.
Leggi anche:
Cause
Cause di immunodeficienza primaria
Questi disturbi possono essere causati da mutazioni, talvolta di un gene in particolare. Se la mutazione è presente sul cromosoma X (sessuale), la malattia che ne deriva si definisce legata al cromosoma X ( Pattern di ereditarietà : Ereditarietà di cromosomi legati al cromosoma X). Questo genere di malattie colpisce maggiormente i bambini di sesso maschile. Il 60% circa delle persone con immunodeficienza primaria è maschio.
I disturbi da immunodeficienza sono classificati in base all’area interessata del sistema immunitario ( Panoramica sul sistema immunitario):
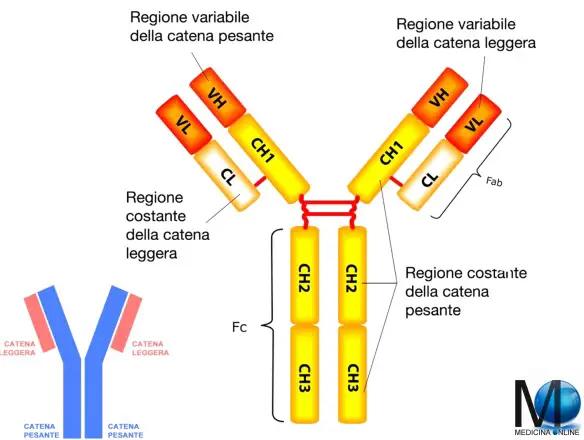
- L’immunità umorale, che coinvolge i linfociti B, un tipo di globuli bianchi che producono anticorpi (immunoglobuline)
- L’immunità cellulare, che coinvolge i linfociti T, un tipo di globuli bianchi che aiutano a identificare e distruggere le cellule estranee o anomale
- Immunità sia umorale sia cellulare
- Fagociti (cellule che ingeriscono e distruggono i microrganismi)
- Proteine del complemento (proteine con svariate funzioni immunitarie, come quella di distruggere i batteri e altre cellule estranee e rendere più facile il riconoscimento e l’ingestione delle cellule estranee da parte di altre cellule del sistema immunitario, Immunità innata : Sistema del complemento).
Il componente interessato del sistema immunitario può essere mancante, ridotto in quantità o alterato e malfunzionante. I problemi legati ai linfociti B sono i più diffusi fra i disturbi da immunodeficienza, rappresentandone oltre la metà.
Alcuni disturbi da immunodeficienza primari:
- Immunodeficienza comune variabile
- Carenza di un anticorpo specifico (immunoglobulina), quale l’IgA
- Ipogammaglobulinemia transitoria infantile
- Agammaglobulinemia legata al cromosoma X
- Immunità cellulare: Malattie dei linfociti T
- Candidosi mucocutanea cronica
- Sindrome di DiGeorge
- Sindrome linfoproliferativa legata al cromosoma X
- Atassia-telangiectasia
- Sindrome da ipergammaglobulinemia E
- Immunodeficienza combinata grave
- Sindrome di Wiskott-Aldrich
- Malattia granulomatosa cronica
- Sindrome di Chédiak-Higashi
- Neutropenia ciclica
- Deficit di adesione leucocitaria
- Carenza dell’inibitore del componente del complemento 1 (C1) (angioedema ereditario)
- Carenza di C3
- Carenza di C5, C6, C7, C8 e/o C9
Patologie da immunodeficienza secondaria
Queste malattie derivano frequentemente dall’uso di farmaci (in particolare gli immunosoppressori). Questi farmaci vengono somministrati al fine di sopprimere il sistema immunitario. Ad esempio, alcuni vengono usati per prevenire il rigetto di un organo o di un tessuto trapiantato ( Farmaci utilizzati per prevenire il rigetto del trapianto). Possono essere somministrati a soggetti con una malattia autoimmune per sopprimere l’attività di attacco dell’organismo nei confronti dei propri tessuti. I corticosteroidi, un tipo di immunosoppressori, sono usati per sopprimere l’infiammazione dovuta a svariate cause, come l’artrite reumatoide. Tuttavia, gli immunosoppressori compromettono anche la capacità dell’organismo di combattere le infezioni e probabilmente di distruggere le cellule tumorali.
La chemioterapia e la radioterapia possono anch’esse sopprimere il sistema immunitario, talvolta causando disturbi da immunodeficienza.
Alcuni farmaci che possono causare immunodeficienza:
- Anticonvulsivanti (usati per il trattamento delle convulsioni)
- Carbamazepina
- Fenitoina
- Valproato
- Immunosoppressori (farmaci che sopprimono il sistema immunitario)
- Azatioprina
- Ciclosporina
- Micofenolato mofetile
- Sirolimus
- Tacrolimus
- Corticosteroidi
- Metilprednisolone
- Prednisone
- Alemtuzumab
- Busulfano
- Ciclofosfamide
- Melfalan
- Adalimumab
- Etanercept
- Infliximab
- Muromonab (OKT3)
- Rituximab
- Tocilizumab
Malattie e condizioni che possono causare immunodeficienza:
- Anemia aplastica
- Leucemia
- Mieloma multiplo, un tipo di tumore
- Anemia a cellule falciformi
- Alcuni tipi di tumore
- Sindrome di Down
- Infezioni da citomegalovirus
- Infezioni da virus di Epstein-Barr
- Infezione da HIV (virus dell’immunodeficienza umana)
- Morbillo
- Varicella
- Alcune infezioni batteriche
- Diabete mellito
- Accumulo di sostanze tossiche nel sangue (uremia)
- Malattia renale cronica
- Sindrome nefrosica
- Epatite
- Insufficienza epatica
- Artrite reumatoide
- Lupus eritematoso sistemico (lupus)
- Asportazione della milza
- Alcolismo
- Ustioni
- Denutrizione
I disturbi da immunodeficienza possono essere indotti quasi da qualsiasi disturbo grave che persiste per lungo tempo. Ad esempio, il diabete può indurre un disturbo da immunodeficienza, poiché la funzionalità dei globuli bianchi è ridotta in presenza di livelli elevati di glicemia. Il virus dell’immunodeficienza umana (HIV) induce sindrome da immunodeficienza acquisita (AIDS), il disturbo da immunodeficienza acquisita più grave e più diffuso.
La denutrizione, generale o legata alla mancanza di specifici nutrienti, può compromettere il sistema immunitario. Quando la denutrizione riduce il peso di una persona pari a meno dell’80% rispetto a quello raccomandato, il sistema immunitario è solitamente compromesso. Un calo pari a meno del 70% determina in genere una grave compromissione.
Leggi anche:
Sintomi
I soggetti con disturbi da immunodeficienza tendono a sviluppare infezioni ripetute. Generalmente, le prime a comparire sono le infezioni respiratorie (come le infezioni dei seni paranasali e dei polmoni) che si ripresentano spesso. La maggior parte dei soggetti sviluppa infine infezioni batteriche gravi che persistono, ricorrono o portano a complicanze. Ad esempio, mal di gola e sindromi da raffreddamento, possono complicarsi sfociando in una polmonite. Tuttavia, la ricorrenza di molti raffreddori non presuppone necessariamente un disturbo da immunodeficienza.
Sono frequenti le infezioni di bocca, occhi e apparato digerente. Il mughetto, un’infezione micotica della bocca, può essere un segno precoce di disturbo da immunodeficienza. Possono comparire ulcerazioni buccali. Possono insorgere gengiviti croniche e infezioni dell’orecchio e della pelle frequenti. Le infezioni batteriche (ad esempio da stafilococchi,) possono causare ulcere piene di pus (pioderma). La pelle dei soggetti affetti da disturbi da immunodeficienza può essere ricoperta da grosse verruche molto visibili, causate da virus.
Molte persone hanno febbre e brividi e perdono l’appetito e/o calano di peso.
Può comparire dolore addominale, eventualmente causato dall’ingrossamento del fegato o della milza.
I neonati o i bambini nella prima infanzia possono avere diarrea cronica e un ritardo dello sviluppo e della crescita (crescita stentata). L’immunodeficienza può essere aggravata se i sintomi si sviluppano nella prima infanzia e non più avanti nel corso della vita.
Altri sintomi variano in base alla gravità e alla durata delle infezioni.
Le immunodeficienze primarie possono verificarsi come parte di una sindrome che presenta altri sintomi, spesso più facili da riconoscere rispetto a quelli dell’immunodeficienza. Ad esempio, un medico potrebbe riconoscere la sindrome di DiGeorge perché i neonati che ne sono affetti hanno una malformazione dei padiglioni auricolari, mandibola piccola con recessione e occhi distanti.
Leggi anche:
- Nove cose che non sai sul tuo sistema immunitario
- Malattie reumatiche: cosa sono, come si curano, sono pericolose?
- Lupus eritematoso sistemico (LES): cause, sintomi e terapie
- Sclerodermia: cause, sintomi e cura
- Sindrome di Sjögren: sintomi, invalidità, terapia e mortalità
- Fibromialgia: sintomi, cause, cura e tender points
- Fibromialgia: dove si trovano i tender points che provocano dolore alla palpazione?
- Dita ippocratiche congenite e secondarie: cause, sintomi e terapie
- Fenomeno di Raynaud: cause, sintomi e trattamento
Diagnosi
Il medico valuta per prima cosa la presenza di un disturbo da immunodeficienza. Esegue esami per l’identificazione di un’anomalia specifica del sistema immunitario.
L’immunodeficienza viene sospettata quando il soggetto presenta infezioni ricorrenti (tipicamente sinusite, bronchite, infezioni dell’orecchio medio o polmonite). I medici sospettano un disturbo da immunodeficienza nel caso di ricorrenza di un’infezione rara o grave o quando un organismo che solitamente non causa gravi infezioni (come funghi Pneumocystis o citomegalovirus) diventa patogeno.
I risultati dell’esame obiettivo possono indicare un’immunodeficienza e, talvolta, il tipo di malattia specifica. Ad esempio, il medico sospetta un certo tipo di disturbo da immunodeficienza quando i linfonodi e le tonsille sono insolitamente piccoli e altri tipi quando questi risultano gonfi e dolorabili alla palpazione.
Per identificare il tipo di disturbo da immunodeficienza, il medico chiede l’età all’insorgenza delle infezioni ricorrenti o rare o altri sintomi caratteristici. I vari tipi di immunodeficienza dipendono principalmente dall’età di insorgenza, come segue:
- Neonati prima dei 6 mesi d’età: generalmente un’anomalia dei linfociti T
- Da 6 a 12 mesi: possibile malattia dei linfociti B e T
- Oltre i 12 mesi: generalmente un’anomalia dei linfociti B e della produzione anticorpale
Il tipo di infezione, potrebbe anche aiutare a identificare il tipo di disturbo da immunodeficienza. Ad esempio, può essere utile individuare l’organo affetto (orecchie, polmoni, cervello o vescica), l’organismo infettante (batteri, funghi o virus) e la specie di quest’ultimo.
I medici s’informano sui fattori di rischio, come diabete, uso di alcuni farmaci, esposizione a sostanze tossiche e sull’eventualità che il paziente abbia parenti stretti con disturbo da immunodeficienza (anamnesi familiare). Si chiedono informazioni sull’attività sessuale precedente e attuale e all’uso di droghe somministrate per via endovenosa, per stabilire se la patologia sia dovuta a infezione da HIV ( Infezione da virus dell’immunodeficienza umana (HIV)).
Esami
Sono necessari esami di laboratorio per confermare la diagnosi e identificare il tipo di disturbo da immunodeficienza. Si preleva un campione di sangue che viene analizzato per determinare il numero totale dei globuli bianchi e le percentuali di ciascuna classe di essi. I globuli bianchi vengono esaminati al microscopio alla ricerca di anomalie. Vengono determinati anche i livelli di immunoglobuline, il numero di globuli rossi e piastrine e i livelli di alcuni tipi di anticorpi prodotti dopo la somministrazione di vaccini. In presenza di risultati alterati vengono eseguiti ulteriori esami.
Qualora si ritenga che l’immunodeficienza sia dovuta a un’anomalia dei linfociti T, vengono eseguiti test cutanei. Il test cutaneo assomiglia al test cutaneo alla tubercolina, usato come controllo della tubercolosi. Piccole quantità di proteine derivanti da organismi infettivi comuni, come il lievito, vengono iniettate sotto pelle. In caso di reazione (eritema, calore e tumefazione) nel giro di 48 ore, i linfociti T funzionano normalmente. L’assenza di una reazione può indicare un’anomalia dei linfociti T. Oppure possono essere ricercate anomalie dei linfociti T eseguendo esami del sangue per stabilire il numero di linfociti T e valutarne la funzionalità.
I soggetti appartenenti a famiglie portatrici di un gene che induce un disturbo da immunodeficienza ereditario possono richiedere di essere sottoposti a un test genetico, per ricercare la presenza di tale gene ed essere informati sul rischio di avere un figlio affetto da tali disturbi. Può essere utile consultare un genetista prima di eseguire i test. Diversi disturbi da immunodeficienza, come l’agammaglobulinemia legata al cromosoma X, la sindrome di Wiskott-Aldrich, l’immunodeficienza combinata grave e la malattia granulomatosa cronica, possono essere riscontrati in un feto, prelevando un campione di liquido attorno al feto (liquido amniotico) o il sangue del feto (test prenatale, Metodi). Questi test sono consigliati ai soggetti con un’anamnesi familiare di disturbi da immunodeficienza, se la mutazione è stata identificata all’interno della famiglia. Alcuni esperti raccomandano di sottoporre a screening tutti i neonati eseguendo un esame del sangue che stabilisce la presenza di linfociti T anomali o scarsi in quantità: si tratta del test TREC (T-cell receptor excision circles). Grazie a questo test è possibile identificare le immunodeficienze cellulari, come l’immunodeficienza combinata grave. L’identificazione precoce di questa patologia nei neonati può prevenirne la morte in tenera età. Il test TREC su tutti i neonati è ora obbligatorio in molti Stati statunitensi.
Leggi anche:
- Shock settico e sepsi: sintomi, terapia, conseguenze, si può guarire
- Differenza tra sepsi e Sindrome da risposta infiammatoria sistemica (SIRS)
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Endocardite: cause, sintomi, diagnosi e terapie
- Differenza tra batteri Gram negativi e Gram positivi
- Differenza tra batteri bacilli, cocchi, streptococchi e spirilli
- Triade di Virchow: i tre fattori di rischio per la trombosi
- Chetosi: cos’è, da cosa è causata, sintomi e terapia in adulti e bambini
- Coagulazione intravascolare disseminata: cause e trattamenti
- Sepsi: cause, sintomi, diagnosi e terapie
- Differenza tra sepsi e setticemia
- Sindrome da disfunzione multiorgano: cause, sintomi, stadi e cure
- Coprocoltura feci per salmonella: perché e come si fa
- Batteriemia: cura, segni, sintomi, diagnosi ed antibiotici
- I cinque segni cardinali dell’infiammazione
- Differenza tra infezione ed infiammazione: sono la stessa cosa?
- Infiammazione: le alterazioni dei vasi sanguigni, permeabilità vascolare e migrazione leucocitaria
- Differenza tra infezione acuta e cronica
- Morte cellulare: differenza tra necrosi, apoptosi ed autofagia
- Infestazione: cos’è, da cosa è causata, come si cura
- Differenza tra infezione ed infestazione
- Differenza tra infestazione interna ed esterna
- Differenza tra infiammazione cronica granulomatosa e non granulomatosa
- Differenza tra granulomi asettici (da corpo estraneo) e settici
- Linfonodi: cosa sono, come riconoscerli, quando sono pericolosi
- Linfonodo sentinella: cos’è e perché è importante in caso di cancro
- Biopsia del linfonodo sentinella: a che serve, perché è importante
- Sistema linfatico e linfonodi: anatomia e funzioni in sintesi
- Differenza tra cisti e linfonodo
- Differenza tra cisti, pseudocisti, ascesso ed empiema
Prevenzione e trattamento
Alcune delle malattie che possono causare immunodeficienza possono essere prevenute e/o trattate, contribuendo a prevenire così lo sviluppo dell’immunodeficienza. Di seguito alcuni esempi:
Infezione da HIV: il rischio d’infezione da HIV può essere ridotto con rapporti sessuali protetti ed evitando di scambiare gli aghi con cui si iniettano le droghe. Anche i farmaci antiretrovirali possono generalmente trattare l’infezione da HIV in maniera efficace ( Infezione da virus dell’immunodeficienza umana (HIV) : Trattamento).
Cancro: il trattamento corretto ripristina di norma la funzionalità del sistema immunitario, a meno che il soggetto non continui ad assumere immunosoppressori.
Diabete: un buon controllo del diabete può contribuire a migliorare la funzione dei globuli bianchi, ostacolando pertanto l’insorgenza delle infezioni.
Le strategie volte alla riduzione del rischio e al trattamento delle infezioni dipendono dal tipo di disturbo da immunodeficienza. Ad esempio, i soggetti con disturbo da immunodeficienza dovuto a carenza di anticorpi sono predisposti alle infezioni batteriche. Quanto segue può contribuire a ridurre il rischio:
- Trattamento periodico con immunoglobuline (anticorpi ottenuti dal sangue di soggetti con sistema immunitario sano), somministrate per via endovenosa o sottocutanea
- Mantenere una buona igiene personale (compresa un’attenta igiene orale)
- Non consumare cibi crudi
- Non bere acqua potenzialmente contaminata
- Evitare il contatto con persone infette
Si somministrano antibiotici alla comparsa di febbre o di un altro segno di infezione, prima di eseguire tecniche chirurgiche e odontoiatriche che possano introdurre batteri nel flusso sanguigno. Se una malattia (come l’immunodeficienza combinata grave) aumenta il rischio di sviluppare infezioni gravi o particolari, è necessario somministrare antibiotici come prevenzione.
I farmaci antivirali vengono somministrati al primo segno di infezione se il soggetto è affetto da un disturbo da immunodeficienza che aumenta il rischio di infezioni virali (come l’immunodeficienza dovuta a un’alterazione dei linfociti T). Questi farmaci comprendono amantadina per l’influenza e aciclovir per l’herpes o la varicella.
Se il disturbo da immunodeficienza specifico non influisce sulla produzione di anticorpi, vengono somministrati i vaccini. I vaccini vengono somministrati per stimolare l’organismo a produrre anticorpi che riconoscono e attaccano batteri e virus specifici. Se il sistema immunitario del soggetto non riesce a produrre anticorpi, la somministrazione di un vaccino non ne promuove la produzione e può addirittura scatenare una malattia. Ad esempio, se una malattia non influisce sulla produzione di anticorpi, la persona affetta da quella malattia deve sottoporsi a vaccino anti-influenzale una volta all’anno. Questo vaccino può anche essere somministrato ai familiari più prossimi del soggetto e alle persone che vivono a stretto contatto con lui/lei. In generale, i vaccini a base di virus vivi non vengono somministrati alle persone con alterazioni dei linfociti B o T perché potrebbero scatenare un’infezione. I vaccini a base di virus vivi includono quello contro rotavirus, morbillo-orecchioni-rosolia, varicella, varicella-zoster (fuoco di S. Antonio), bacillo di Calmette-Guérin (bacille Calmette-Guérin, BCG) e influenza, somministrato come spray nasale. Un vaccino vivo per il virus della poliomielite somministrato per via orale non viene più utilizzato negli Stati Uniti, ma lo è ancora in alcune parti del mondo.
Il trapianto delle cellule staminali ( Trapianto di cellule staminali) può correggere alcuni disturbi da immunodeficienza, soprattutto l’immunodeficienza combinata grave. Le cellule staminali vengono solitamente prelevate dal midollo osseo, ma talvolta dal sangue (compreso quello prelevato dal cordone ombelicale). Il trapianto delle cellule staminali, disponibile in alcuni centri medici importanti, è solitamente riservato ai soggetti con patologie gravi.
Talvolta è utile eseguire il trapianto del tessuto timico. La terapia genica per alcune patologie congenite da immunodeficienza è stata sperimentata con successo.
Grazie al trattamento adeguato, molti soggetti affetti da immunodeficienza possono godere di un’aspettativa di vita normale. Alcuni, tuttavia, necessitano di cure intensive e frequenti per tutta la vita. Altri, come quelli affetti da immunodeficienza combinata grave, muoiono durante l’infanzia, a meno che non ricevano un trapianto di midollo osseo o di cellule staminali.
Leggi anche:
- Sistema immunitario, immunità innata e specifica: riassunto, schema e spiegazione
- Immunità innata (aspecifica): barriere, infiammazione e complemento
- Immunità innata (aspecifica): neutrofili, macrofagi e linfociti natural killer
- Immunità specifica (acquisita): linfociti, T killer, T helper, T γδ, B ed anticorpi
- Immunità specifica (acquisita): memoria passiva, attiva ed immunizzazione
- Immunità specifica (acquisita) umorale e cellulare
- Patologie del sistema immunitario: immunodeficienze, autoimmunità ed ipersensibilità
- Anticorpi: (immunoglobuline): tipi, caratteristiche e funzioni
- Differenza tra antigene, aptene allergene ed epitopo
- Aptene: cos’è e perché è importante per il sistema immunitario
- Antigene: cos’è e perché è importante per il sistema immunitario
- Differenza tra antigeni esogeni, endogeni, tumorali, nativi ed autoantigeni
- Epitopi sequenziali e conformazionali: cosa sono e come funzionano
- HIV: dopo quanto si manifestano i sintomi? I 4 stadi dell’infezione
- Differenza tra HIV e AIDS: sono uguali?
- Si muore di AIDS? Qual è l’aspettativa di vita?
- HIV: sintomi iniziali in donne e uomini
- Differenza tra malattia, sindrome e disturbo con esempi
- Differenza tra virus HIV1 e HIV2
- Sesso e AIDS: l’HIV si trasmette anche tramite il rapporto orale
- Il liquido pre-eiaculatorio può indurre gravidanza e trasmettere l’HIV?
- HIV e AIDS: come, dove e quando si eseguono i test per la diagnosi?
- Invasività microbica: la capacità di invadere l’organismo ospite
- Differenza tra invasività clinica e microbica
- Il virus più pericoloso del mondo è più vicino a te di quanto pensi
- Qual è il virus che ha ucciso più persone in assoluto?
- Qual è il virus più letale al mondo?
- Quante persone uccide ogni anno il virus HIV che causa l’AIDS?
- L’insospettabile influenza ogni anno uccide più persone dell’Ebola
- Il virus che uccide mezzo milione di bambini ogni anno
- Febbre gialla, dengue ed altre malattie trasmesse dalle zanzare
- Febbre dengue: sintomi, trasmissione, diagnosi, terapia e prevenzione
- Virus mortali: ecco gli 11 più pericolosi al mondo
- Sierotipo in microbiologia: significato ed importanza per i vaccini
- Differenza tra DNA ed RNA
- Differenza tra organismi autotrofi ed eterotrofi
- Differenza tra organismi prototrofi ed auxotrofi
- Differenza tra organico inorganico
- I 12 batteri più pericolosi per l’uomo
- Differenza tra microrganismi, batteri, virus, microbi e germi
- Differenza tra funghi, muffe e lieviti
- Differenza tra capside a simmetria icosaedrica, elicoidale e complessa
- Differenza tra unicellulare e pluricellulare con esempi
- Tubercolosi: trasmissione, sintomi, diagnosi e cure in sintesi
- Tubercolosi: cause e patogenesi della malattia
- Mycobacterium tuberculosis: il batterio che causa la tubercolosi
- Sintomi della tubercolosi polmonare ed extrapolmonare
- Come si trasmette la tubercolosi?
- Tubercolosi: diagnosi e progressione della malattia
- Test cutaneo della tubercolina: Test di Mantoux per la tubercolosi
- Trattamento farmacologico per la tubercolosi
- Trattamento della tubercolosi resistente ai farmaci
- Tubercolosi: prognosi, vaccino e strategie di prevenzione
- Papilloma vescicale: virus, sintomi, vaccino e cure
- Virus del papilloma (HPV): tipi più pericolosi ed a basso rischio
- Come si contrae il Virus del papilloma (HPV)?
- Infezione da Virus del papilloma, gravidanza e problemi al feto
- Trattamento del Virus del papilloma (HPV)
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
Condividi questo articolo:

 Il sistema immunitario è un insieme di processi notevolmente efficace che incorpora specificità, inducibilità e adattamento. Tuttavia possono verificarsi dei malfunzionamenti ed essi si possono dividere in tre grandi categorie: immunodeficienze, malattie autoimmuni e ipersensibilità.
Il sistema immunitario è un insieme di processi notevolmente efficace che incorpora specificità, inducibilità e adattamento. Tuttavia possono verificarsi dei malfunzionamenti ed essi si possono dividere in tre grandi categorie: immunodeficienze, malattie autoimmuni e ipersensibilità.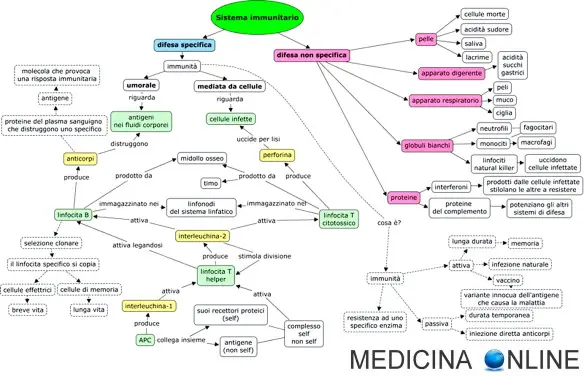 Il sistema immunitario è una complessa rete integrata di mediatori chimici e cellulari, di strutture e processi biologici, sviluppatasi nel corso dell’evoluzione, per difendere l’organismo da qualsiasi forma di insulto chimico, traumatico o infettivo alla sua integrità. Per funzionare correttamente, un sistema immunitario deve essere in grado di rilevare un’ampia varietà di agenti, noti come agenti patogeni, dai virus agli elminti e distinguerli dal proprio tessuto sano dell’organismo.
Il sistema immunitario è una complessa rete integrata di mediatori chimici e cellulari, di strutture e processi biologici, sviluppatasi nel corso dell’evoluzione, per difendere l’organismo da qualsiasi forma di insulto chimico, traumatico o infettivo alla sua integrità. Per funzionare correttamente, un sistema immunitario deve essere in grado di rilevare un’ampia varietà di agenti, noti come agenti patogeni, dai virus agli elminti e distinguerli dal proprio tessuto sano dell’organismo. Il mieloma multiplo è un tumore che colpisce le plasmacellule, una componente molto importante del sistema immunitario. In particolare le plasmacellule sono il risultato della maturazione dei linfociti B che, assieme ai linfociti T, rappresentano le due principali tipologie cellulari coinvolte nella risposta immunitaria. Il ruolo delle plasmacellule, che si trovano soprattutto nel midollo osseo, è quello di produrre e liberare anticorpi per combattere le infezioni, ma in alcuni casi la loro crescita procede in maniera incontrollata dando origine al tumore.
Il mieloma multiplo è un tumore che colpisce le plasmacellule, una componente molto importante del sistema immunitario. In particolare le plasmacellule sono il risultato della maturazione dei linfociti B che, assieme ai linfociti T, rappresentano le due principali tipologie cellulari coinvolte nella risposta immunitaria. Il ruolo delle plasmacellule, che si trovano soprattutto nel midollo osseo, è quello di produrre e liberare anticorpi per combattere le infezioni, ma in alcuni casi la loro crescita procede in maniera incontrollata dando origine al tumore.