
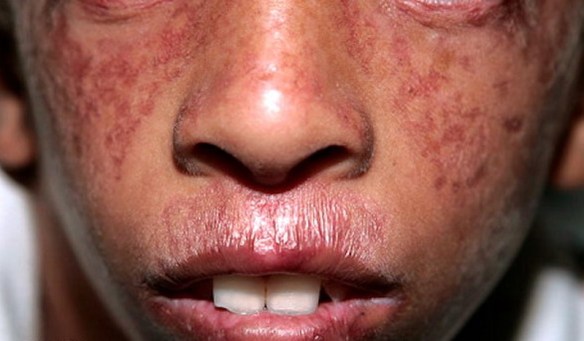
 La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie.
La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie.
Segni e sintomi
Le tipiche lesioni cutanee sono teleangiectasiche eritematose, appaiono in infanzia al volto, con tipico aspetto a farfalla, ma occasionalmente si riscontrano anche ai dorsi delle mani e dei piedi. Queste aree sono sensibili al sole, e peggiorano dopo l’esposizione. L’ altra caratteristica fondamentale della malattia e la bassa statura, soprattutto prenatale all’esordio, ma che persiste anche in infanzia e in eta adulta. La maggior parte degli individui rimane al di sotto dei 148 cm.
I maschi sono sterili con azospermia, mentre le femmine possono essere fertili.
Altre caratteristiche sono: macchie caffelatte, microcefalia, dolicocefalia, faccia stretta, naso e/o orecchie prominenti, la tipica tonalita alta della voce, l’ipertrigliceridemia. L’intelligenza e normale. Le infezioni sono frequenti, a causa dell’immunodeficienza, e possono essere minacciose per la vita.
Leggi anche:
- Le malattie genetiche più diffuse al mondo
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
Storia naturale
La sindrome di Bloom e un disordine genetico che predispone al cancro. Infatti, quasi la meta dei pazienti sviluppano almeno una neoplasia (10% dei quali hanno piu di una neoplasia primitiva, il che e proprio tipico di questa sindrome); l’eta media di insorgenza del primo tumore e di 25 anni (range 2-49 anni). Dopo i trent’anni si manifestano leucemie acute (ALL e ANLL) nel 15% dei casi e linfomi pure nel 15% dei casi. Carcinomi (una grande varieta di tipi) si sviluppano nel 30% dei casi, perlopiu dopo i 20 anni.
A parte le malattie neoplastiche, le complicanze maggiori dal punto di vista medico sono la malattia polmonare cronica e il diabete mellito (nel 10% dei pazienti).
L’incidenza della sindrome e di circa 2/100.000 nati tra gli Ebrei Ashkenazi e tra i Giapponesi (effetto del fondatore: le persone affette derivano da un antenato comune); per il resto e molto rara.
Eziologia
La sindrome di Bloom e trasmessa con modalita autosomica recessiva. Il gene e stato mappato: location 15q26.
Diagnosi
Fa diagnosi il riscontro (patognomonico) di un alto numero di scambi spontanei di cromatidi fratelli (90 per cellula, piu di 10 volte maggiore che nel normale); in alcuni pazienti, il riscontro di una sottopopolazione con basso numero di scambi suggerisce una ricombinazione tra alleli materni e paterni (con mutazioni differenti), che da origine ad un gene selvaggio funzionante. Gli eterozigoti, invece, non si possono investigare con studi citogenetici.
Terapia
Non esistono cure definitive. L’ormone della crescita è controindicato nelle sindromi in cui vi e un aumentato rischio di rotture cromosomiche, come appunto la sindrome di Bloom. Controlli frequenti assicurano la diagnosi precoce dei tumori associati alla sindrome di Bloom. Gli individui affetti da questa patologia devono evitare i raggi X, gli agenti chemioterapici ed altre esposizioni ambientali che possono provocare danni ai loro cromosomi particolarmente fragili.
Leggi anche:
- Cos’è un cromosoma ed a che serve?
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Cos’è un gene ed a che serve?
- Cosa sono gli alleli ed a che servono?
- Differenza tra cellule eucariote e procariote
- Virus e virioni: cosa sono, come sono fatti, come funzionano e come si riproducono
- Differenza tra cellula aploide e diploide con esempi
- Riproduzione cellulare e ciclo cellulare
- Meiosi: spiegazione di tutte tappe
- Mitosi: spiegazione delle quattro fasi
- Differenza tra mitocondri e cloroplasti
- Differenza tra citosol e citoplasma
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Organelli (organuli) citoplasmatici della cellula animale: cosa sono ed a che servono?
- Mitocondri: definizione, dimensioni e funzioni
- Citoscheletro: funzioni e struttura
- Ribosomi e reticolo endoplasmatico: cosa sono e che funzioni svolgono?
- Nucleo cellulare: funzioni, dimensioni e membrane nucleari
- Lisosomi: cosa sono? Significato e dimensioni
- Perossisomi: definizione e funzioni
- Membrana plasmatica: definizione e funzioni
- Apparato del Golgi: spiegazione semplice e funzioni
- Citosol: definizione e funzioni
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
 Il lupus eritematoso sistemico (da cui l’acronimo “LES“, o semplicemente “lupus“; in inglese “systemic lupus erythematosus“, da cui l’acronimo “SLE“) è una patologia del connettivo cronica di natura autoimmune, che può colpire diversi organi e tessuti del corpo. Come accade nelle altre malattie autoimmuni, il sistema immunitario produce autoanticorpi che, invece di proteggere il corpo da virus, batteri e agenti estranei, aggrediscono cellule e componenti del corpo stesso, causando infiammazione e danno tissutale. Il meccanismo patogenetico è un’ipersensibilità di III tipo, caratterizzata dalla formazione di immunocomplessi. Il lupus è una patologia caratterizzata da manifestazioni eritematose cutanee e mucose, sensibilità alla luce del sole e coinvolgimento sistemico di quasi tutti gli organi e apparati come il rene, le articolazioni, il sistema nervoso centrale, le sierose e il sistema emopoietico, dovute a deposito di immunocomplessi e complemento. Colpisce più frequentemente le donne, soprattutto fra i 15 e i 40 anni.
Il lupus eritematoso sistemico (da cui l’acronimo “LES“, o semplicemente “lupus“; in inglese “systemic lupus erythematosus“, da cui l’acronimo “SLE“) è una patologia del connettivo cronica di natura autoimmune, che può colpire diversi organi e tessuti del corpo. Come accade nelle altre malattie autoimmuni, il sistema immunitario produce autoanticorpi che, invece di proteggere il corpo da virus, batteri e agenti estranei, aggrediscono cellule e componenti del corpo stesso, causando infiammazione e danno tissutale. Il meccanismo patogenetico è un’ipersensibilità di III tipo, caratterizzata dalla formazione di immunocomplessi. Il lupus è una patologia caratterizzata da manifestazioni eritematose cutanee e mucose, sensibilità alla luce del sole e coinvolgimento sistemico di quasi tutti gli organi e apparati come il rene, le articolazioni, il sistema nervoso centrale, le sierose e il sistema emopoietico, dovute a deposito di immunocomplessi e complemento. Colpisce più frequentemente le donne, soprattutto fra i 15 e i 40 anni.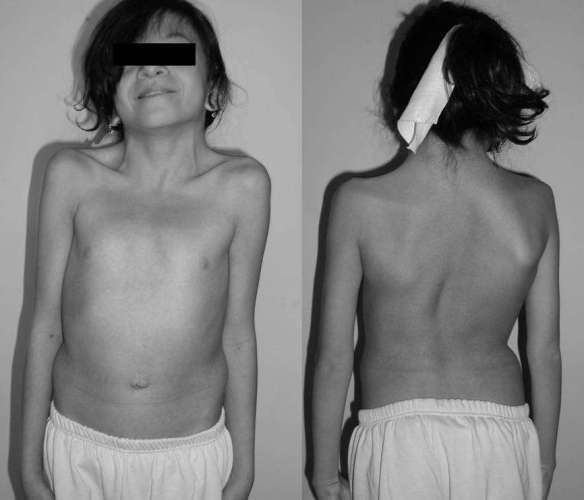 La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto).
La sindrome di Noonan è una malattia genetica dalle caratteristiche cliniche particolarmente variabili, con un’incidenza stimata di un caso ogni 1000-2500 nati. Le manifestazioni più frequenti includono cardiopatie congenite (nel 70% dei pazienti), bassa statura, un aspetto caratteristico del volto (abbassamento delle palpebre, occhi distanziati, orecchie grandi e ruotate posteriormente), malformazioni del torace, deficit cognitivi variabili, tendenza al sanguinamento e criptorchidismo (mancata discesa dei testicoli nello scroto). Il melograno (Punica granatum) è una pianta appartenente alla famiglia delle Punicaceae, originario dell’Asia sud-occidentale ed ormai diffuso in tutto il mondo. Il frutto del melograno si chiama granata o melagrana. Con il termine melograno spesso si indicano sia l’albero che il frutto, ma più correttamente in italiano il frutto viene chiamato melagrana, tuttavia da ora in poi nell’articolo chiamerò il frutto con “melograno”. Il nome melograno deriva dal latino malum (“mela”) e granatum (“con semi”). La forma del melograno ricorda in effetti quella di una mela, ma ecco all’interno la sorpresa dei suoi numerosi chicchi dal gusto leggermente acidulo.
Il melograno (Punica granatum) è una pianta appartenente alla famiglia delle Punicaceae, originario dell’Asia sud-occidentale ed ormai diffuso in tutto il mondo. Il frutto del melograno si chiama granata o melagrana. Con il termine melograno spesso si indicano sia l’albero che il frutto, ma più correttamente in italiano il frutto viene chiamato melagrana, tuttavia da ora in poi nell’articolo chiamerò il frutto con “melograno”. Il nome melograno deriva dal latino malum (“mela”) e granatum (“con semi”). La forma del melograno ricorda in effetti quella di una mela, ma ecco all’interno la sorpresa dei suoi numerosi chicchi dal gusto leggermente acidulo. Scoperti i geni che ci fanno scegliere i cibi: si aprono così le porte alle diete “genetiche” personalizzate e alla nascita di cibi “dietetici ma buoni”. Una serie di scoperte italiane dimostra infatti che la predilezione per i cibi, dall’amore per la pancetta all’odio per i broccoli, si nasconde nel Dna. Lo si deve a stato un gruppo di ricerca dell’Università di Trieste e Istituto di Ricovero e Cura a Carattere Scientifico (Irccs) Burlo Garofolo coordinato da Paolo Gasparini, che ha presentato i risultati raggiunti in occasione della conferenza della European Society of Human Genetics (Eshg).
Scoperti i geni che ci fanno scegliere i cibi: si aprono così le porte alle diete “genetiche” personalizzate e alla nascita di cibi “dietetici ma buoni”. Una serie di scoperte italiane dimostra infatti che la predilezione per i cibi, dall’amore per la pancetta all’odio per i broccoli, si nasconde nel Dna. Lo si deve a stato un gruppo di ricerca dell’Università di Trieste e Istituto di Ricovero e Cura a Carattere Scientifico (Irccs) Burlo Garofolo coordinato da Paolo Gasparini, che ha presentato i risultati raggiunti in occasione della conferenza della European Society of Human Genetics (Eshg). Il futuro della ricerca medica è in larga parte legato alle conoscenze sul genoma umano, che via via si accumulano. La sfida in atto riguarda però la possibilità di incidere direttamente sul Dna modificando i geni all’interno di una singola cellula.
Il futuro della ricerca medica è in larga parte legato alle conoscenze sul genoma umano, che via via si accumulano. La sfida in atto riguarda però la possibilità di incidere direttamente sul Dna modificando i geni all’interno di una singola cellula. Un salumiere che tocca le fette di prosciutto che vi sta incartando al supermercato e – con le stesse mani – manipola i vostri soldi: una scena che dovrebbe spingervi a
Un salumiere che tocca le fette di prosciutto che vi sta incartando al supermercato e – con le stesse mani – manipola i vostri soldi: una scena che dovrebbe spingervi a  Immaginate di soffrire il mal di denti ma di non provare alcun dolore. Oppure di ricevere un pugno sul naso e continuare senza problemi a fare quel che stavate facendo.
Immaginate di soffrire il mal di denti ma di non provare alcun dolore. Oppure di ricevere un pugno sul naso e continuare senza problemi a fare quel che stavate facendo.