
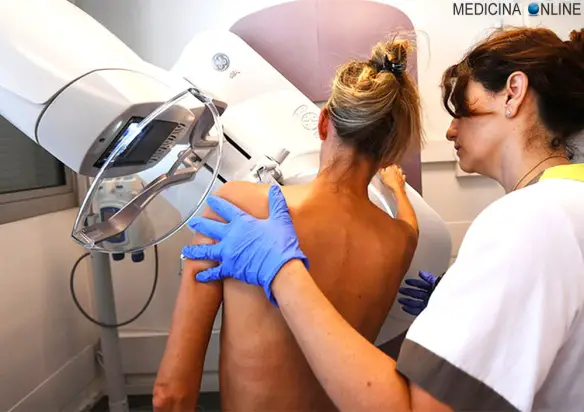
 I BRCA (Breast Related Cancer Antigens) 1 e 2, sono due geni che intervengono nel controllo del ciclo cellulare e codificano per la “proteina per la suscettibilità al carcinoma mammario”. Una loro mutazione aumenta di circa 8 volte il rischio di cancro alla mammella. I BRCA sono diventati “famosi” presso il grande pubblico nel 2013 per via della doppia mastectomia a cui si è sottoposta la famosa attrice statunitense Angelina Jolie. L’attrice ha infatti dichiarato di avere la mutazione del gene BRCA1 e secondo i medici aveva l’87% di rischio di sviluppare il cancro alla mammella e il 50% di rischio di sviluppare il cancro alle ovaie, dato che la madre, la nonna e la zia erano morte per la stessa causa e che ora, grazie all’asportazione delle mammelle, le probabilità sono calate al 5%.
I BRCA (Breast Related Cancer Antigens) 1 e 2, sono due geni che intervengono nel controllo del ciclo cellulare e codificano per la “proteina per la suscettibilità al carcinoma mammario”. Una loro mutazione aumenta di circa 8 volte il rischio di cancro alla mammella. I BRCA sono diventati “famosi” presso il grande pubblico nel 2013 per via della doppia mastectomia a cui si è sottoposta la famosa attrice statunitense Angelina Jolie. L’attrice ha infatti dichiarato di avere la mutazione del gene BRCA1 e secondo i medici aveva l’87% di rischio di sviluppare il cancro alla mammella e il 50% di rischio di sviluppare il cancro alle ovaie, dato che la madre, la nonna e la zia erano morte per la stessa causa e che ora, grazie all’asportazione delle mammelle, le probabilità sono calate al 5%.
Un nuovo studio
Un nuovo studio sembra però confermare che mutazione del BRCA, ormai soprannominato “gene Jolie”, non è la condanna a morte che prima si pensava, dato che se si ha un tumore al seno le probabilità di sopravvivenza sono le stesse rispetto alle pazienti che non hanno il DNA mutato. Lo studio è stato eseguito dall’università di Southampton e pubblicato dalla rivista Lancet Oncology, che ha anche trovato che la mastectomia dopo la diagnosi non ha effetti sulla speranza di sopravvivenza.
Mortalità uguale
Lo studio ha esaminato i dati di 2733 donne tra i 18 e i 40 anni che avevano avuto una diagnosi di tumore al seno, di cui il 12% aveva la mutazione. A dieci anni dalla diagnosi non erano sopravvissute al cancro 651 donne, e la mortalità è risultata uguale in entrambi i gruppi. Un terzo delle donne con la mutazione aveva optato per la doppia mastectomia, sottolineano gli autori, ma questo tipo di intervento non ha cambiato la probabilità di sopravvivenza. “Questo ci dice che l’intervento radicale non deve essere fatto subito, insieme agli altri trattamenti – sottolinea alla BBC Diane Eccles, l’autore principale -, anche se probabilmente la mastectomia può dare benefici a lungo termine, venti o trent’anni dopo la diagnosi iniziale”.
Leggi anche:
- Cancro al seno: sintomi precoci, diagnosi, terapia e prevenzione
- Biopsia del linfonodo sentinella: a che serve, perché è importante
- La mammografia: un esame rapido che può salvarti la vita
- L’ecografia mammaria: un esame innocuo ed indolore che ti può salvare vita
- Capezzolo retratto (introflesso): cause, cancro al seno e trattamenti
- Tumore alla mammella: cause, diagnosi e prevenzione
- Riconoscere il cancro al seno: sintomi precoci e tardivi
- Tumore al seno: stadiazione, prognosi e sopravvivenza
- Cancro al seno: metastasi e sintomi avanzati del tumore
- Differenza tra ecografia e mammografia nella diagnosi di tumore al seno
- Noduli al seno: quando preoccuparsi ed andare dal medico?
- Come riconoscere un nodulo maligno del seno da uno benigno?
- Tumore al seno: sintomi e dolore al braccio
- Tumore al seno C1 C2 C3 C4 C5: cosa significa il referto?
- Tumore al seno età: a quanti anni si può verificare?
- Classificazione e stadiazione delle fasi del tumore alla mammella
- Capezzolo dolorante e sensibile in uomo, donna, gravidanza e menopausa
- Malattia di Paget del capezzolo: sintomi precoci, cause e cure
- Mammella: anatomia e funzioni del seno e delle ghiandole mammarie
- Divisione in quadranti della mammella (Q1 Q2 Q3 Q4)
- Quadranti mammari, tumore al seno, quadrantectomia e mastectomia radicale
- Breast Unit salvavita: -18% di mortalità in caso di cancro al seno
- La visita senologica: come, quando e perchè farla?
- Differenza dei capezzoli e del seno in gravidanza
- A cosa serve e come si fa l’autopalpazione del seno?
- Autopalpazione della mammella: come farla nel modo giusto [VIDEO]
- Perché ho il seno piccolo? Quali sono i fattori che influenzano la grandezza del mio seno?
- Ginecomastia: quando è l’uomo ad avere il seno
- Differenza tra ginecomastia vera, falsa, acquisita, congenita e puberale
- L’uomo può allattare al seno?
- Storia di un seno: dall’embrione alla menopausa
- Cos’è la pubertà, a che età inizia e come si manifesta?
- Il reggiseno provoca il cancro al seno? Finalmente abbiamo una risposta
- Perché agli uomini piace così tanto il seno delle donne?
- Ormoni estrogeni: cosa sono e quali funzioni svolgono?
- Differenza dolore al seno da gravidanza e da ciclo mestruale
- Differenze tra neonati allattati con latte materno ed artificiale
- Quando una mammella non si sviluppa: la Sindrome di Poland
- Politelia: quando i capezzoli sono troppi, cause e terapie
- Mastodinia: quando il seno è gonfio e dolorante
- Rassodare il seno senza chirurgia con la Radiofrequenza Monopolare
- Aumentare il seno senza chirurgia con i cibi ricchi di fitoestrogeni
- Vuoi avere un seno più bello? Comincia con la postura e la ginnastica giusta
- Dimagrire senza perdere seno
- Prendere il sole al seno fa male? Come abbronzarlo in sicurezza
- Grandezza del seno: le donne del nord battono quelle del sud
- La donna con il seno più grande del mondo [VIDEO]
- I deodoranti che contengono alluminio causano il cancro al seno?
- Esercizi e consigli per rassodare e tonificare il seno
- Il reggiseno che può far crescere il tuo seno
- L’incidenza del tumore al seno in crescita tra le quarant’enni: l’importanza della mammografia
- Linfonodo sentinella: cos’è e perché è importante in caso di cancro
- Rossore ed irritazione della pelle sotto e tra il seno: cause e rimedi
- Fitoestrogeni: rimedi naturali in menopausa
- Ingrandire il seno con l’ipnosi
- Filler ad alta densita’: Macrolane™ per l’aumento del seno senza bisturi
- I fitoestrogeni aumentano il rischio di cancro al seno?
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
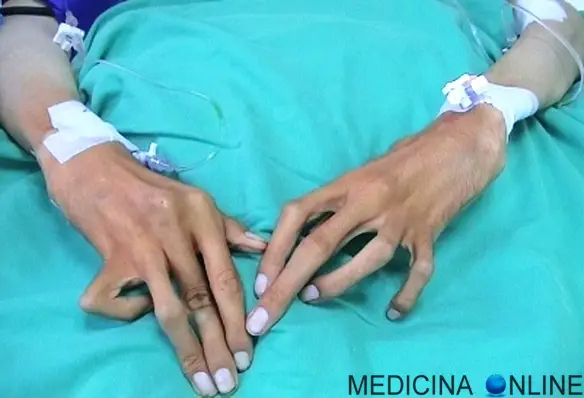 Con “aracnodattilia” (“dita a ragno”) si intende una deformità dello scheletro delle mani caratterizzata da dita affusolate e sproporzionatamente allungate rispetto al palmo della mano o alla pianta del piede. Queste estremità possono risultare, inoltre, insolitamente flessibili, similmente alle zampe di un ragno, motivo per cui questa patologia è così chiamata. Costituisce uno dei sintomi della Sindrome di Marfan ed è caratterizzata dal “segno del pollice” e dal “segno del polso”. L’aracnodattilia può essere congenita e svilupparsi nel contesto di diverse malattie genetiche. Altre persone possono manifestare questo allungamento patologico delle dita delle mani e dei piedi nel tempo, durante l’infanzia o l’età adulta.
Con “aracnodattilia” (“dita a ragno”) si intende una deformità dello scheletro delle mani caratterizzata da dita affusolate e sproporzionatamente allungate rispetto al palmo della mano o alla pianta del piede. Queste estremità possono risultare, inoltre, insolitamente flessibili, similmente alle zampe di un ragno, motivo per cui questa patologia è così chiamata. Costituisce uno dei sintomi della Sindrome di Marfan ed è caratterizzata dal “segno del pollice” e dal “segno del polso”. L’aracnodattilia può essere congenita e svilupparsi nel contesto di diverse malattie genetiche. Altre persone possono manifestare questo allungamento patologico delle dita delle mani e dei piedi nel tempo, durante l’infanzia o l’età adulta.
 Il momento in cui sarà possibile scegliere l’altezza, il colore degli occhi o il quoziente intellettivo di un figlio è davvero molto vicino, almeno in base ad un articolo pubblicato sulla rivista del MIT, Technology Review, in cui viene citata una compagnia statunitense che pianifica di fornire una analisi genetica preimpianto sugli embrioni che vada oltre le malattie.
Il momento in cui sarà possibile scegliere l’altezza, il colore degli occhi o il quoziente intellettivo di un figlio è davvero molto vicino, almeno in base ad un articolo pubblicato sulla rivista del MIT, Technology Review, in cui viene citata una compagnia statunitense che pianifica di fornire una analisi genetica preimpianto sugli embrioni che vada oltre le malattie. Impariamo da bambini che il il cuore si trova a sinistra nel nostro petto, ma vi siete mai chiesto il perché di questa posizione? Un gruppo di ricercatori dell’Istituto di Neuroscienze di Alicante ha provato a dare una risposta a questa domanda, grazie ad una ricerca pubblicata su Nature.
Impariamo da bambini che il il cuore si trova a sinistra nel nostro petto, ma vi siete mai chiesto il perché di questa posizione? Un gruppo di ricercatori dell’Istituto di Neuroscienze di Alicante ha provato a dare una risposta a questa domanda, grazie ad una ricerca pubblicata su Nature.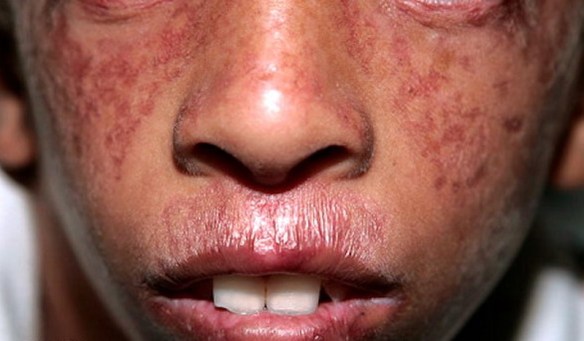
 La fibrosi cistica polmonare o, più semplicemente, “fibrosi cistica” (acronimo FC, in inglese “cystic fibrosis”) è una rara, debilitante e
La fibrosi cistica polmonare o, più semplicemente, “fibrosi cistica” (acronimo FC, in inglese “cystic fibrosis”) è una rara, debilitante e 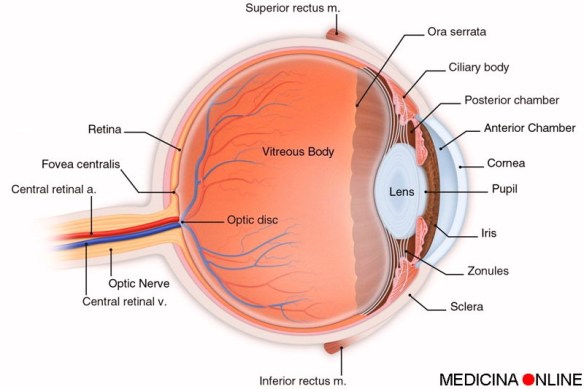 La maculopatia, o degenerazione maculare, è un termine che racchiude diverse patologie degenerative che danneggiano progressivamente la macula, cioè la zona più ricca di fotorecettori che si trova al centro della retina e che permette di distinguere i dettagli più fini delle immagini. Le parti più esterne della retina ci permettono invece di vedere tutto ciò che si trova intorno al punto che stiamo fissando. Per questo motivo, anche nei casi più gravi, la degenerazione maculare non provoca cecità totale perché la visione periferica e laterale viene conservata. La maculopatia può essere principalmente di due tipi: “secco” od “umido”.
La maculopatia, o degenerazione maculare, è un termine che racchiude diverse patologie degenerative che danneggiano progressivamente la macula, cioè la zona più ricca di fotorecettori che si trova al centro della retina e che permette di distinguere i dettagli più fini delle immagini. Le parti più esterne della retina ci permettono invece di vedere tutto ciò che si trova intorno al punto che stiamo fissando. Per questo motivo, anche nei casi più gravi, la degenerazione maculare non provoca cecità totale perché la visione periferica e laterale viene conservata. La maculopatia può essere principalmente di due tipi: “secco” od “umido”.