
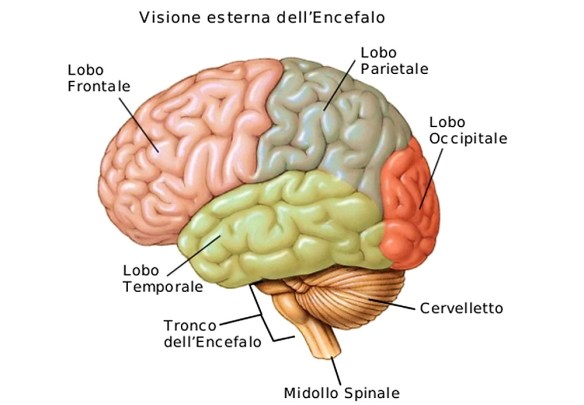
 La malattia (o morbo o còrea) di Huntington è una patologia ereditaria causata dalla degenerazione di neuroni situati in specifiche aree cerebrali – striato e corteccia cerebrale – e caratterizzata da una generale atrofia del cervello. I sintomi iniziali possono essere bruschi mutamenti dell’umore, apatia, irritabilità, depressione e rabbia, difficoltà nella guida, nell’imparare cose nuove o nel prendere una decisione. Altri possono presentare cambiamenti nella scrittura e movimenti involontari delle dita, dei piedi, del viso o del tronco (chiamati “còrea” dal termine greco che significa “danza”). In altri casi possono insorgere disturbi dell’equilibrio e del coordinamento motorio con accentuato rischio di cadute. L’ordine di comparsa di questi sintomi e la gravità possono variare notevolmente da un individuo all’altro, così come l’età d’insorgenza.
La malattia (o morbo o còrea) di Huntington è una patologia ereditaria causata dalla degenerazione di neuroni situati in specifiche aree cerebrali – striato e corteccia cerebrale – e caratterizzata da una generale atrofia del cervello. I sintomi iniziali possono essere bruschi mutamenti dell’umore, apatia, irritabilità, depressione e rabbia, difficoltà nella guida, nell’imparare cose nuove o nel prendere una decisione. Altri possono presentare cambiamenti nella scrittura e movimenti involontari delle dita, dei piedi, del viso o del tronco (chiamati “còrea” dal termine greco che significa “danza”). In altri casi possono insorgere disturbi dell’equilibrio e del coordinamento motorio con accentuato rischio di cadute. L’ordine di comparsa di questi sintomi e la gravità possono variare notevolmente da un individuo all’altro, così come l’età d’insorgenza.
Età di insorgenza
Nella sua forma più classica la malattia insorge tra i 35 e i 45 anni, ma in alcuni casi può manifestarsi prima dei 20 anni con un rapido declino del rendimento scolastico/lavorativo, cambiamenti nella scrittura, rigidità, tremori, lentezza e rapidi spasmi muscolari. La forma giovanile progredisce molto più rapidamente di quella adulta. La forma tardiva, infine, si manifesta dopo i 55 anni e può essere più complessa da diagnosticare per la compresenza di altre patologie, ma anche perché i sintomi possono essere particolarmente lievi e perciò più difficili da individuare.
Diffusione
La frequenza della malattia è stimata in 5-10/100.000 individui, ma non sempre viene correttamente diagnosticata: esiste perciò certamente un gran numero di portatori inconsapevoli.
Come si trasmette la malattia di Huntington?
La malattia si trasmette con modalità autosomico dominante: un genitore affetto ha cioè una probabilità del 50% di trasmetterla a ciascuno dei suoi figli, a prescindere dal sesso. Quello che viene trasmesso non è la malattia, ma la mutazione di un gene, IT-15, localizzato sul cromosoma 4 e codificante per la proteina huntingtina, dalle funzioni non ancora del tutto note ma comunque importante per lo sviluppo embrionale e nel cervello adulto. Il gene IT-15 sano presenta al suo interno una specifica sequenza CAG che si ripete fino a un massimo di 35 volte, ma che in presenza della mutazione si ripete per un numero eccessivo di volte (da 39 a oltre 100). Ogni individuo che eredita il gene mutato svilupperà più o meno precocemente la malattia.
Come avviene la diagnosi della malattia di Huntington?
Dal 1993, con la scoperta del gene responsabile, è disponibile il test genetico per diagnosticare la malattia di Huntington. Su un prelievo di sangue di un individuo a rischio ma ancora del tutto privo di sintomi è possibile condurre un’analisi genetica che consente di accertare la presenza o l’assenza della mutazione nel gene IT-15. La decisione di sottoporsi al test è molto delicata, perché il risultato coinvolge non solo il singolo individuo, ma anche la sua famiglia (coniuge, figli, fratelli). È perciò consigliabile fare riferimento a centri di ricerca che dispongono di professionisti idonei. Data l’attuale impossibilità di prevenire la malattia non vengono effettuati test sui minori di 18 anni. Inoltre la diagnosi corretta si basa non solo sul test genetico, ma anche sulla storia familiare del paziente e sulle immagini del cervello che si possono ottenere con TC e risonanza magnetica nucleare: vista la variabilità della sintomatologia, anche in base alla fase e alla durata della malattia, è fondamentale considerare le informazioni fornite dai vari strumenti disponibili e non da un solo elemento (per esempio un paziente in fase iniziale può avere TC e risonanza magnetica normali).
Terapie
Purtroppo non esiste al momento una terapia risolutiva. I principali sintomi motori e psichiatrici possono essere tenuti sotto controllo con farmaci in grado di migliorare la qualità di vita del malato e prevenire eventuali complicazioni, ma non di guarire la malattia o interromperne il decorso. Sono tuttavia in corso diverse sperimentazioni cliniche per valutare l’efficacia di varie sostanze.
Leggi anche:
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Distrofia muscolare di Duchenne, di Becker e di Emery-Dreifuss
- Distrofia muscolare in adulti e bambini: sintomi, cause, diagnosi e cure
- Distrofia miotonica di tipo 1 e 2: cause, trasmissione, sintomi, diagnosi, cure
- Le malattie genetiche più diffuse al mondo
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Differenza tra distrofia muscolare e sclerosi multipla
- Differenza tra distrofia muscolare e SLA (sclerosi laterale amiotrofica)
- Cos’è un cromosoma ed a che serve?
- Differenza tra allele dominante e recessivo
- Differenza tra omozigote ed eterozigote
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome del tramonto o del crepuscolo: cause, sintomi e cura
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Che cos’è l’intelligenza umana: definizione, significato e psicologia
- Quoziente d’intelligenza: valori, significato, test ed ereditarietà
- Problem solving: cos’è, caratteristiche, tecniche, fasi ed esempi
- Sindrome di Tourette: cause, sintomi, diagnosi e trattamento
- Sindrome di Tourette: si può guarire definitivamente? Come si guarisce?
- Differenza tra gene e allele
- Differenza tra genotipo e fenotipo
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Cos’è un gene ed a che serve?
- Cosa sono gli alleli ed a che servono?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
 Esistono diversi modi per classificare il dolore. Uno di questi consiste nel differenziarlo base alla sua durata e alla sua ripetibilità. Prendiamo ad esempio un tipico dolore molto diffuso e conosciuto nella popolazione generale: la lombalgia.
Esistono diversi modi per classificare il dolore. Uno di questi consiste nel differenziarlo base alla sua durata e alla sua ripetibilità. Prendiamo ad esempio un tipico dolore molto diffuso e conosciuto nella popolazione generale: la lombalgia. HIV e AIDS non sono affatto la stessa cosa, anche se ovviamente sono termini tra loro legati. Cerchiamo di fare un po’ di chiarezza.
HIV e AIDS non sono affatto la stessa cosa, anche se ovviamente sono termini tra loro legati. Cerchiamo di fare un po’ di chiarezza.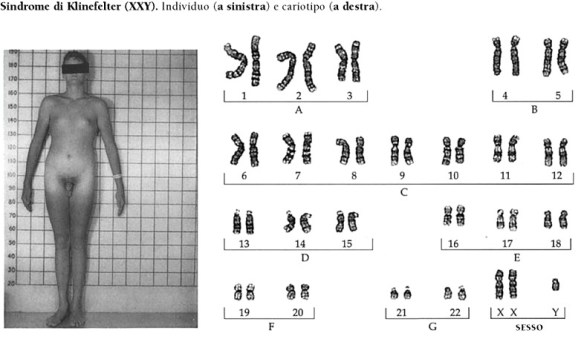 La sindrome di Klinefelter è una condizione caratterizzata dalla presenza di un cromosoma sessuale X in più nei soggetti di sesso maschile (47,XXY). Questa condizione prende il nome dal medico statunitense Harry Klinefelter, del Massachusetts General Hospital di Boston, che nel 1942 pubblicò i risultati delle sue ricerche su nove uomini che manifestavano testicoli ipotrofici, aumento del volume delle mammelle e diminuzione/mancanza di peli sulla superficie corporea. L’assetto cromosomico dei soggetti aventi tali caratteristiche fisiche fu identificato negli anni Cinquanta; negli anni Settanta, l’istituto statunitense National Institute of Child Health and Human Development avviò uno screening su larga scala per determinare il tipo di cromosomi sessuali presenti in 40.000 neonati, e la frequenza della condizione XXY. Attualmente, sembra che l’incidenza di XXY sia relativamente alta (circa uno su 1000 neonati maschi); in realtà, solo una bassa percentuale di questi individui sviluppano una vera e propria sindrome, cioè un insieme di disturbi correlati al loro particolare assetto cromosomico. Per questo motivo, molti autori hanno preferito abbandonare la vecchia denominazione di sindrome di Klinefelter, e indicano i soggetti in questione semplicemente come “maschi-XXY”.
La sindrome di Klinefelter è una condizione caratterizzata dalla presenza di un cromosoma sessuale X in più nei soggetti di sesso maschile (47,XXY). Questa condizione prende il nome dal medico statunitense Harry Klinefelter, del Massachusetts General Hospital di Boston, che nel 1942 pubblicò i risultati delle sue ricerche su nove uomini che manifestavano testicoli ipotrofici, aumento del volume delle mammelle e diminuzione/mancanza di peli sulla superficie corporea. L’assetto cromosomico dei soggetti aventi tali caratteristiche fisiche fu identificato negli anni Cinquanta; negli anni Settanta, l’istituto statunitense National Institute of Child Health and Human Development avviò uno screening su larga scala per determinare il tipo di cromosomi sessuali presenti in 40.000 neonati, e la frequenza della condizione XXY. Attualmente, sembra che l’incidenza di XXY sia relativamente alta (circa uno su 1000 neonati maschi); in realtà, solo una bassa percentuale di questi individui sviluppano una vera e propria sindrome, cioè un insieme di disturbi correlati al loro particolare assetto cromosomico. Per questo motivo, molti autori hanno preferito abbandonare la vecchia denominazione di sindrome di Klinefelter, e indicano i soggetti in questione semplicemente come “maschi-XXY”.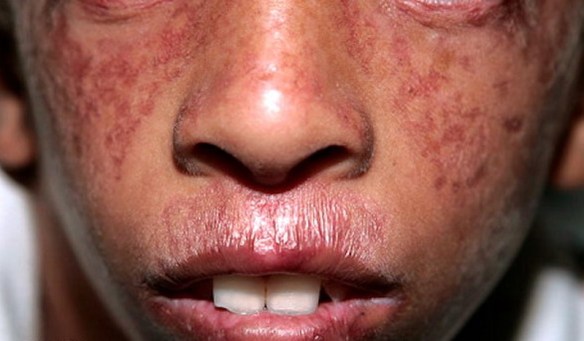 La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie.
La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie.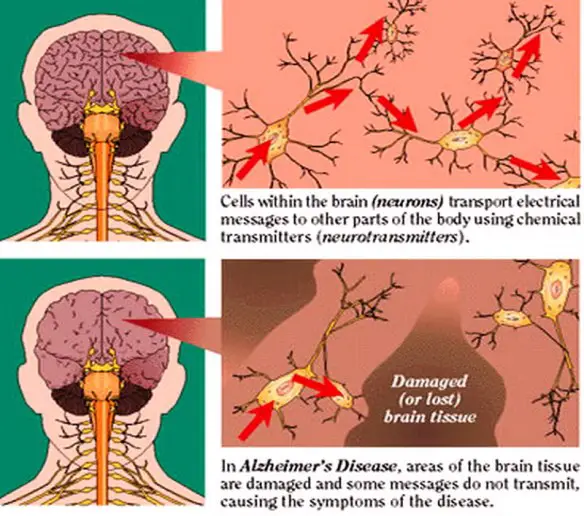 La causa per la maggior parte dei casi di Alzheimer è ancora in gran parte sconosciuta, ad eccezione che per casi dall’1% al 5% in cui sono state individuate le differenze genetiche esistenti.
La causa per la maggior parte dei casi di Alzheimer è ancora in gran parte sconosciuta, ad eccezione che per casi dall’1% al 5% in cui sono state individuate le differenze genetiche esistenti. Il giorno in cui è morto, David Kirby aveva 32 anni. Giaceva sul letto, il suo volto sofferente e il corpo consumato dall’Aids, circondato dalla sua famiglia. Quell’istante in cui l’uomo è spirato, è stato impresso dalla macchina fotografica di Therese Frare, all’epoca una giovane studentessa di foto-giornalismo in un’università dell’Ohio. Frare riuscì a catturare quel momento straziante con freddezza e professionalità, dando un volto umano a quella malattia fino ad allora socialmente stigmatizzata. Nel novembre del 1990, la rivista Life decise di pubblicare quella foto che divenne rapidamente l’immagine simbolo per tutte le persone affette dal virus dell’Hiv.
Il giorno in cui è morto, David Kirby aveva 32 anni. Giaceva sul letto, il suo volto sofferente e il corpo consumato dall’Aids, circondato dalla sua famiglia. Quell’istante in cui l’uomo è spirato, è stato impresso dalla macchina fotografica di Therese Frare, all’epoca una giovane studentessa di foto-giornalismo in un’università dell’Ohio. Frare riuscì a catturare quel momento straziante con freddezza e professionalità, dando un volto umano a quella malattia fino ad allora socialmente stigmatizzata. Nel novembre del 1990, la rivista Life decise di pubblicare quella foto che divenne rapidamente l’immagine simbolo per tutte le persone affette dal virus dell’Hiv.
