
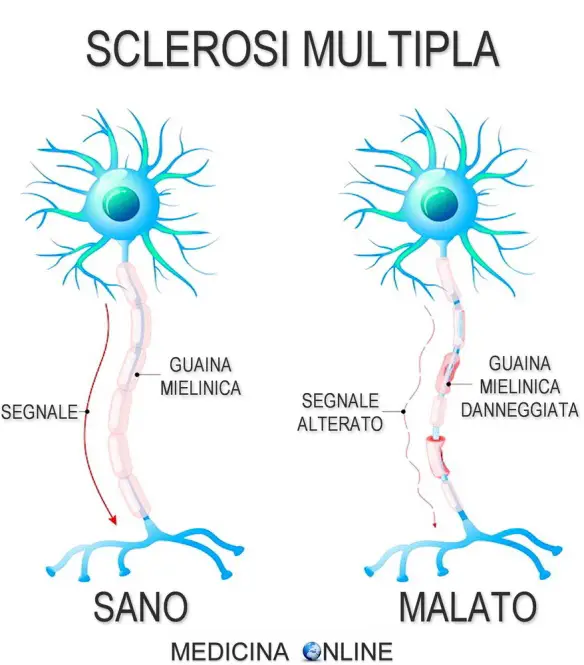
 In clinica il termine “demielinizzante” si usa per indicare una patologia che interessi soprattutto le guaine mieliniche delle fibre nervose, lasciando relativamente intatti gli assoni e le loro cellule di origine. Altri connotati istopatologici di un vero processo demielinizzante sono l’assenza di degenerazione walleriana secondaria (per la relativa assenza di compromissione dei cilindrassi), un’infiltrazione di cellule infiammatorie a livello perivascolare e – spesso – la localizzazione perivenosa della demielinizzazione.
In clinica il termine “demielinizzante” si usa per indicare una patologia che interessi soprattutto le guaine mieliniche delle fibre nervose, lasciando relativamente intatti gli assoni e le loro cellule di origine. Altri connotati istopatologici di un vero processo demielinizzante sono l’assenza di degenerazione walleriana secondaria (per la relativa assenza di compromissione dei cilindrassi), un’infiltrazione di cellule infiammatorie a livello perivascolare e – spesso – la localizzazione perivenosa della demielinizzazione.
Le malattie elencate nella tabella che segue sono conformi a quest’ultima definizione e tutte condividono un’altra caratteristica, che è quella di avere una eziologia non ancora del tutto nota, ma presumibilmente autoimmune:
Classificazione delle malattie demielinizzanti
- Sclerosi multipla
- Forma encefalomielopatica cronica
- Sclerosi multipla acuta
- Neuromielite ottica (Malattia di Devic)
- Sclerosi cerebrale diffusa (encefalite periassiale diffusa) di Schilder e sclerosi concentrica di Balò
- Encefalomielite e mielite acuta disseminata (post-infezioni)
- Conseguente a infezione da EBV (virus di Epstein-Barr), CMV (citomegalovirus), herpesvirus, Mycoplasma o un’altra infezione non definita
- Conseguente a morbillo, varicella, vaiolo e, raramente, parotite, rosolia e influenza o un’altra infezione di natura non determinata
- Conseguente a vaccinazione antirabbica o antivaiolosa
- Encefaliti emorragiche necrotizzanti acute e subacute
- Forma encefalopatica acuta (leucoencefalite emorragica di Hurst)
- E. Mielopatia necrotizzante subacuta.
Tra queste affezioni, la più importante è di gran lunga la sclerosi multipla. È omesso da questa classificazione un gruppo di malattie che comprende la degenerazione subacuta combinata dovuta a deficit di vitamina B12, la leucoencefalopatia multifocale progressiva e la demielinizzazione corticale dell’encefalopatia ipossica – ciascuna caratterizzata da demielinizzazione marcata, ma con un fattore causale ben definito e unico.
In questo articolo ci occuperemo in particolare della sclerosi multipla, dal punto di vista epidemiologico e delle manifestazioni cliniche.
Sclerosi multipla
La sclerosi multipla (da cui l’acronimo SM) è una malattia del sistema nervoso centrale (SNC) che inizia per lo più nella tarda adolescenza e nella prima età adulta e si manifesta con attacchi distinti, ricorrenti, di alterata funzionalità del midollo spinale, del tronco encefalico, del nervo ottico e del cervello. La lesione sottostante è caratterizzata da distruzione della mielina, ma anche gli assoni e altre strutture possono essere coinvolte. Gli attacchi hanno un’insorgenza subacuta, ma possono essere acuti e sono spesso seguiti da remissione dei sintomi e persino da guarigione. In molti casi il decorso recidivante-remittente assume impercettibilmente le caratteristiche di un quadro cronico progressivo.
Epidemiologia
La distribuzione geografica della malattia è un elemento importante. Nelle zone settentrionali degli Stati Uniti, in Canada, in Gran Bretagna e nel nord dell’Europa la prevalenza è alta: da 30 a 80 per 100000. Nelle regioni meridionali dell’Europa e degli Stati Uniti la prevalenza scende a 6-14 per 100000 e nelle aree equatoriali a meno di 1 per 100000. Si ritiene che gli individui che emigrano da un’area ad alto rischio a una a
basso rischio (o viceversa) dopo i 15 anni di età mantengano il valore di rischio tipico della loro zona di origine. Se l’emigrazione avviene prima di questa età, essi acquisiscono il rischio caratteristico della zona in cui emigrano. L’incidenza familiare è bassa, ma per i parenti di primo grado di un soggetto affetto il rischio è comunque più elevato rispetto alla popolazione generale. Alcuni antigeni di istocompatibilità (HLA) sono più frequenti nei soggetti con SM (HLA-DR2, -DR3, -B7 e A3). Raramente la SM si manifesta nei bambini. Le donne adulte sono più suscettibili degli uomini (1,7:1) e i soggetti di razza bianca più di quelli di razza nera. Né il trauma né la gravidanza sembrano essere [attori eziopatogenetici.
Sintomi e segni
Raramente la malattia ha inizio in forma asintornatica e in questi casi la si riscontra soltanto accidentalmente a una risonanza magnetica (RM) eseguita per altre cause. Generalmente il primo attacco si verifica senza preavviso e può essere mono- o polisintomatico. In un quinto dei casi l’insorgenza è acuta, cioè il deficit raggiunge la sua massima gravità nell’arco di minuti od ore. I sintomi di esordio più comuni, che si manifestano in varie combinazioni, sono un’ipostenia o parestesie a un arto, perdita monoculare della vista, diplopia, vertigini, ipostenia o parestesie facciali, atassia e nistagmo. Nella maggior parte dei casi al primo attacco fa seguito una remissione. Gli attacchi successivi costituiscono prevalentemente una riacutizzazionedelle lesioni precedenti, oppure possono essere l’espressione di nuove lesioni. In un periodo di tempo variabile, di solito anni, la disabilità del paziente aumenta progressivamente; egli può presentare, in varie combinazioni, una paraparesi asimmetrica e segni di interessamento del fascio corticospinale; atassia sensitiva e cerebellare, incontinenza urinaria, atrofia ottica, nìstagmo, oftalmoplegia intemucleare e disartria. Solo nel 3-4% dei pazienti si verificano crisi epilettiche. Le turbe psichiche sono variabili, in relazione al prevalere di lesioni spinali o cerebrali e al numero di queste ultime. Lo stadio tardivo stabilizzato può essere raggiunto non prima che siano passati 20 o 25 anni dall’esordio. Una volta raggiunto lo stadio avanzato, il deterioramento può essere così lento da suggerire la presenza di una patologia degenerativa. Altri pazienti, invece, peggiorano rapidamente, entro 3 o 4 anni, e in rari casi il paziente può arrivare al decesso alcuni mesi dopo l’insorgenza (SM acuta). Può verificarsi anche una progressione lenta della malattia senza episodi di ricaduta, specialmente in età più avanzata. Non si registrano segni sistemici, a parte l’affaticabilità.
Neurite ottica retrobulbare
Una forma particolare di malattia demielinizzante coinvolge il nervo ottico (che è un’estensione del sistema nervoso centrale) e risulta essere la manifestazione iniziale della SM quasi nel 25% dei pazienti. Nell’arco di parecchie ore o di alcuni giorni si sviluppano una macchia monoculare di visione offuscata o cecità, dolore all’occhio nei movimenti del globo oculare e una desaturazione del colore rosso. La papilla ottica può apparire normale (neurite retrobulbare) o edematosa (papillite), a seconda del punto di lesione nel nervo; il riflesso pupillare risulta alterato. Metà o più dei pazienti in cui la malattia esordisce esclusivamente con neurite ottica presenterà altri sintomi di SM, generalmente dopo alcuni anni ma talvolta dopo molti anni dal primo episodio.
La terapia della neurite ottica va praticata con alte dosi di corti costeroidi per via endovenosa, in quanto questi farmaci accelerano la regressione del deficit visivo, anche se probabilmente non modificano l’esito finale, che generalmente è positivo per quanto riguarda la vista; in effetti, un ampio studio ha riscontrato che la somministrazione di steroidi per via orale può persino aumentare la frequenza di ricadute.
Mielite trasversa
Questo termine è talvolta erroneamente utilizzato per descrivere una lesione demielinizzante che coinvolge le strutture del midollo in corrispondenza di uno o più segmenti adiacenti. Si tratta di una delle sindromi di presentazione della SM, ma può riconoscere anche altre cause, tra cui un processo post-infettivo.
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Cos’è la sclerosi multipla e cosa determina? Spiegazione semplice
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Test di Romberg: cos’è, a che serve, come si esegue
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Differenza tra tremori, spasmi, miotonia, crampi, fascicolazioni, tic
- Differenza tra discinesia, ipocinesia, ipercinesia, tardiva e primaria
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Astenia, quando mancano le forze fisiche o mentali: cause, diagnosi, cure
- Nevrastenia (esaurimento nervoso): cause, diagnosi, cure
- Ipostenia (miastenia): cause, sintomi, diagnosi, terapie, complicanze, prognosi
- Ipotonia e atonia: definizione, etimologia, significato, esempi
- Ipotonia muscolare in neonati, adulti, anziani: cause, sintomi, cure, consigli
- Ipotonia nel lattante: cause, sintomi, diagnosi e trattamento
- Iperstenia: definizione, significato, etimologia, fisiologica e patologica
- Ipertonia: definizione, significato, etimologia
- Ipertonia muscolare: cause, tipi, sintomi, diagnosi, terapie
- Cos’è il tono muscolare, a che serve, come mantenerlo?
- Distonia focale e generalizzata: tipi, sintomi, diagnosi e terapie
- Miotonia: cause, sintomi, diagnosi e terapie
- Demenza senile: cause, sintomi, decorso e cure
- Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Mielite: infettiva, cervicale, dorsale, trasversa, si guarisce?
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Segni meningei e irritazione meningi in bambini ed adulti
- Meningite: incubazione, fulminante, sintomi, contagio e cura
- Encefalite: conseguenze, è contagiosa, danni, si guarisce?
- Encefalite autoimmune da anticorpi anti-NMDA: trattamento, sintomi
- Meningismo: triade, segni, cause, diagnosi, definizione e cura
- Differenza tra meningite e meningismo: qual è più grave?
- Differenza tra meningite, encefalite, meningoencefalite, encefalomielite
- Differenza tra meningite virale e batterica
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Puntura lombare: complicanze, risultati, è dolorosa, a che serve?
- Meningi: anatomia, funzioni e patologia in sintesi
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Mielopatia: significato, tipi, sintomi, diagnosi,cure, consigli, prognosi
- Segni meningei e irritazione meningi in bambini ed adulti
- Tumore al cervello: cause, sintomi iniziali e tardivi, diagnosi, cura, sopravvivenza, aspettativa di vita
- Ipertensione endocranica: valori, cause, bradicardia, terapie
- Pseudotumor cerebri (ipertensione endocranica benigna) cause e cure
- Operato al cervello per un tumore mentre suona il clarinetto
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Esoftalmo bi- e monolaterale: cause, gravità, conseguenze e cure
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Sindrome della sella vuota: cause, sintomi, diagnosi e terapie
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Shunt cerebrale e intervento per il drenaggio permanente
- Ventricoli cerebrali: anatomia e funzioni in sintesi
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Emorragia cerebrale: conseguenze, riabilitazione e recupero
- Emorragia subaracnoidea: cause, conseguenze, linee guida
- Ematoma subdurale: cos’è, da quale malattia è provocata, recidivo, decorso
- Coma da emorragia cerebrale: quanto può durare?
- Differenza tra proencefalo, mesencefalo, romboencefalo, telencefalo, diencefalo
- Differenza tra metencefalo e mielencefalo
- Emicrania con aura: cause, sintomi, diagnosi e trattamenti
- Emicrania senza aura: cause, sintomi, diagnosi e trattamenti
- Differenza tra emicrania con aura ed emicrania senza aura
- Cos’è l’aura emicranica?
- Ictus cerebrale emorragico e ischemico: cause, sintomi, diagnosi, cure, rischi
- Differenza tra ictus cerebrale ed attacco ischemico transitorio (TIA)
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Cosa si prova e cosa succede quando si rompe un aneurisma cerebrale?
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Sindrome frontale o ipofrontalità: cause, sintomi, conseguenze, diagnosi, cure
- Malformazioni artero-venose cerebrali: sintomi e cura
- Ischemia: cos’è, cause, conseguenze, rischi, cure
- Necrosi: significato, definizione, sinonimo, cause, cure
- Trombo: cause, classificazione, trombosi venose, arteriose e sistemiche
- Infarto, ischemia, necrosi, aterosclerosi, trombo, embolo, ictus, miocardio… Facciamo chiarezza
- Tronco cerebrale (mesencefalo, ponte e bulbo) anatomia e funzioni in sintesi
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Torcicollo spasmodico e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Disturbi della stazione eretta e della deambulazione in neurologia
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Aprassia ideazionale, ideomotoria e melocinetica: sintomi, diagnosi, cure
- Aprassia: significato, etimologia, tipi, zone del cervello coinvolte
- Aprassia: test usati per la diagnosi e tipici errori aprassici
- Aprassia costruttiva (apractognosia): cause, sintomi, diagnosi, terapie
- Disartria: cause, sintomi, diagnosi, trattamento
- Perdita della coordinazione muscolare: l’atassia
- Differenze tra aprassia, atassia, disprassia, afasia, disartria, anartria
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Anche gli esseri umani possono avere una coda
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Siringobulbia: cause, sintomi, diagnosi e trattamento
- Glasgow Coma Scale per la classificazione del coma
- Pediatric Glasgow Coma Scale in italiano: scala pediatrica del coma
- Differenza tra toracentesi, paracentesi e rachicentesi
- Narcolessia: cause, sintomi, cure e terapia farmacologica
- Catatonia: significato, definizione, cause, sinonimi e cure
- Cataplessia: causa, significato, nel sonno, cura ed etimologia
- Catalessia in medicina: cause, sintomi, nel sonno e cure
- Differenza tra catatonia, catalessia e cataplessia
- Paralisi del sonno e allucinazioni ipnagogiche: cause, pericoli, rimedi
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Differenza tra stato vegetativo, di minima coscienza, coma, sonno e stato soporoso
- Differenza tra sindrome locked-in e stato vegetativo
- Differenza tra stato vegetativo persistente e permanente
- Differenza tra stato vegetativo e stato di minima coscienza
- Commozione cerebrale: cos’è, cosa fare, conseguenze, tempi di recupero
- Differenza tra commozione cerebrale, trauma cranico e contusione cerebrale
- Trauma cranico: ematoma, commotivo, sintomi tardivi, cosa fare
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Poligono di Willis: anatomia e varianti anatomiche
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Differenza tra afasia, disartria ed aprassia
- Area di Broca: funzioni ed afasia di Broca
- Area di Wernicke: funzioni ed afasia di Wernicke
- Differenza tra afasia di Broca e di Wernicke
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- A cosa serve il midollo osseo?
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Si può morire di epilessia?
- Sindrome pseudobulbare: cause, sintomi, diagnosi e terapie
- Malattia di Binswanger: cause, sintomi, diagnosi, cure
- Sistema piramidale e fascio genicolato: anatomia, decorso, funzioni
- Sistema extrapiramidale: anatomia, decorso, vie, funzioni, patologie
- Differenza tra via piramidale ed extrapiramidale
- Miclono: cause, sintomi, caratteristiche, quando preoccuparsi, cure
- Spasmi muscolari e mioclonie: da cosa sono causati?
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Spasmi muscolari e mioclonie: cura, trattamento e rimedi
- Spasmi muscolari e mioclonie: come si fa la diagnosi?
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
