
 Cos’è il DNA
Cos’è il DNA
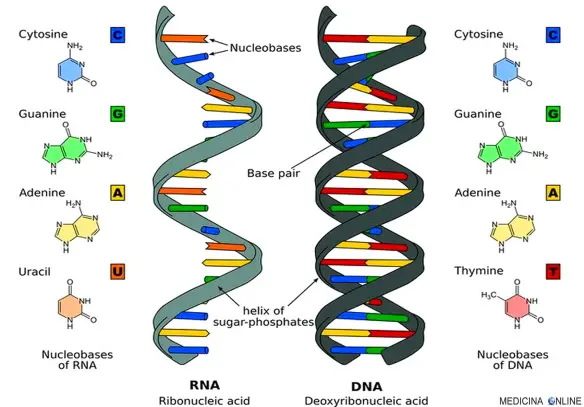
Tutti quanti sappiamo che il DNA racchiude in sé le informazioni genetiche di ciascuno di noi. Come fosse una sorta di memoria fisica su cui sono iscritte le istruzioni di programmazione del software umano. Ma cosa vuol dire questo in termini scientifici? Il termine DNA nasconde la sua stessa natura, perché si traduce in acido deossiribonucleico, che è la composizione chimica di questo acido nucleico. In quanto acido nucleico, si tratta di un polimero formato da unità chiamate nucleotidi, che si organizzano in una struttura complessa. Ogni nucleotide è composto da tre elementi: un gruppo fosfato, uno zucchero (o deossiribosio) e una base azotata. E’ proprio da questa sua natura polinucleotide che deriva la caratteristica doppia elica utilizzata per raffigurare il DNA. Il DNA è, infatti, formato da due catene (conosciute anche come filamenti) di nucleotidi che si avvolgono a spirale l’una intorno all’altra. Grazie a studi effettuati negli anni ’50, sappiamo anche che le due catene sono orientate in senso opposto, ovvero i nucleotidi hanno opposta sequenza di atomi di carbonio del deossiribosio. Il DNA è presente nel nucleo di tutte le cellule di un organismo (che possono essere i cosiddetti cromosomi), sempre uguale a se stesso in quanto porta iscritta la sequenza genetica. Il codice genetico è dato, quindi, dalla peculiare disposizione in sequenza dei nucleotidi, sempre diversa tra gli esseri viventi.
Nel DNA si nasconde il segreto di quello che siamo, in un certo senso, perché è lì che si trovano le istruzioni genetiche che ci hanno fatto proprio come siamo. In concreto, cosa fa il DNA per consentire lo sviluppo di un organismo? Contenendo al suo interno le informazioni genetiche fondamentali, consente la biosintesi dell’RNA e poi delle proteine, che sono poi le molecole necessarie per ogni organismo.
La traduzione dell’mRNA permette la sintesi di una nuova proteina
Cos’è l’RNA
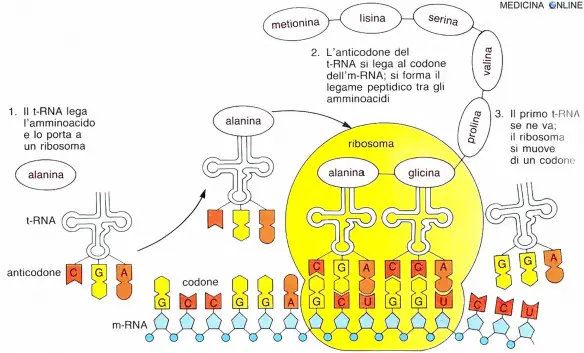
Il passaggio dalle informazioni contenute nel DNA alle proteine non è diretto. Esiste infatti un passaggio intermedio fondamentale che porta dagli amminoacidi alla proteina, attraverso un processo di traduzione genetica noto come sintesi proteica. Questo passaggio è la creazione di una nuova molecola intermedia, una sorta di duplicato del DNA (pur con qualche differenza strutturale). Questa molecola intermedia è, appunto, l’RNA, ovvero acido ribonucleico. Essendo generato per trascrizione dal DNA, dal punto di vista chimico l’RNA è molto simile, anche se consta di un singolo filamento di nucleotidi invece della doppia elica. Per dirla in parole semplici, l’RNA non è altro che il frutto della trascrizione di un filamento di DNA, in cui le informazioni genetiche vengono ricopiate in una nuova molecola (traduzione) per formare una nuova proteina. L’RNA funge da “stampo”. Pur essendo una copia ed essendo presente in ogni cellula, però, si tratta di una molecola molto complessa, che può assumere ruoli differenti nel corso della creazioni dell’organismo. Nel processo di trascrizione genetica, infatti, non vengono creati solo filamenti RNA per la traduzione del codice genetico, ma anche altri che svolgono funzioni biologiche (come i ribosomi). In generale possiamo distinguere tre tipologie di RNA:
- RNA messaggero: contiene l’informazione per la sintesi delle proteine (funge da stampo).
- RNA ribosomiale: che entra nella struttura dei ribosomi.
- RNA transfer: utilizzato per la traduzione nei ribosomi.
Grazie al polimero RNA, quindi, è reso possibile il processo di divisione cellulare e replicazione del DNA, senza che in questo percorso vada persa qualche fondamentale informazione genetica.
Quali sono le differenze?
Avendo descritto le caratteristiche chimiche e funzionali di DNA e RNA, le differenze tra i due dovrebbe essere abbastanza chiaro. Visto che ci troviamo in campi complessi come quelli della chimica e della genetica, però, forse è meglio un riassunto finale.
Partiamo dalle differenze strutturali:
- la composizione è molto simile, catena polinucleotidica contenente quattro nucleotidi diversi. Però il DNA è composto da due catene antiparallele che si avvolgono a elica (doppio filamento), mentre l’RNA è quasi sempre a un solo filamento.
- Nei nucleotidi che si nasconde la differenza chimica fondamentale tra le due molecole. L’RNA contiene uno zucchero diverso (ribosio al posto di deossiribosio) e anche una base diversa (uracile al posto di timina).
Differenza nella funzione:
- Il DNA contiene nel suo interno il codice genetico, ovvero tutte le informazioni necessarie per lo sviluppo delle proteine e, quindi, dell’organismo.
- L’RNA è il frutto di un processo di copia del DNA, così se il DNA resta stabile e immutato nel tempo, i filamenti di RNA hanno utilizzo immediato e svolgono, come abbiamo visto, diverse funzioni nello sviluppo concreto delle proteine. Possiamo dire, per concludere, che il DNA è il calco originale (o il master di un disco) e le molecole RNA che ne derivano sono le copie da utilizzare concretamente.
Leggi anche:
- Differenza tra DNA ed RNA
- Differenza tra DNA, RNA, ADN, ARN, A-DNA, B-DNA, Z-DNA
- Differenze tra mRNA, tRNA e rRNA
- Sintesi proteica: trascrizione, sintesi dell’RNA, RNA polimerasi I, II e III
- Traduzione dell’mRNA e sintesi delle proteine nei ribosomi
- Proteine trasportate nel nucleo, nei mitocondri, nei cloroplasti, nel reticolo endoplasmatico
- Differenza tra tra sintesi proteica, trascrizione e traduzione in genetica
- Differenza tra malattia autosomica dominante e recessiva con esempi
- Differenza tra dominanza semplice, incompleta e codominanza
- Differenza tra allele dominante e recessivo
- Differenza tra cellule eucariote e procariote
- Differenza tra cellula aploide e diploide con esempi
- Ereditarietà X linked, malattie legate al cromosoma X: significato, esempi
- Differenza tra portatore sano e malato in genetica
- Cosa significa “portatore sano” in genetica e nelle infezioni?
- Differenza tra omozigote ed eterozigote
- Differenza tra genotipo e fenotipo
- Variazione del numero dei cromosomi: aneuploidia, monoploidia, poliploidia
- Aneuplodia, nullisomia, monosomia, trisomia, tetrasomia
- Mutazioni cromosomiche: delezione, duplicazione, inversione, traslocazione
- Delezione in genetica: spiegazione semplice ed esempi
- Duplicazione in genetica: spiegazione semplice ed esempi
- Inversione in genetica: spiegazione semplice ed esempi
- Traslocazione in genetica: spiegazione semplice ed esempi
- Le 10 malattie genetiche più diffuse al mondo
- Sindrome di Down: cause, sintomi in gravidanza e nei neonati
- Sindrome di Turner: cariotipo, cause, sintomi e segni caratteristici
- Sindrome di Rett: cause, sintomi, tipi, diagnosi, stadi, cure, morte
- Sindrome di Klinefelter: cariotipo, cause, sintomi e cura
- Sindrome di Asperger in bambini ed adulti: primi sintomi, terapie
- Sindrome di Noonan: cause, sintomi nel neonato, aspettative di vita
- Sindrome di Bloom: cause, sintomi, diagnosi e terapia
- Sindrome di Pfeiffer: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Stickler: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Möbius: cause, sintomi, diagnosi e terapia
- Sindrome di Pierre Robin: cause, sintomi, diagnosi, cure, prognosi
- Sindrome di Wiskott-Aldrich: cause, sintomi, trasmissione, cure
- Sindrome di Lesch-Nyhan: cause, sintomi, trasmissione, diagnosi, cure
- Sindrome di Hughes-Stovin: cause, sintomi, diagnosi e terapia
- Sindrome di Ehlers-Danlos: la malattia dei contorsionisti
- Sindrome di Prader-Willi: cause, trasmissione, sintomi, diagnosi, cure
- Ritardo mentale nei bambini lieve, moderato, grave: si guarisce?
- Sindrome dell’X fragile in uomini e donne: sintomi, aspettativa di vita, cure
- Sindrome dell’idiota sapiente: cause, caratteristiche e sintomi
- Sindrome dell’addome a prugna secca: cause, sintomi, diagnosi, terapie
- Quando una mammella non si sviluppa: la sindrome di Poland
- Diabete insipido: cause, diagnosi e trattamento
- Distrofia muscolare in adulti e bambini: sintomi, cause, diagnosi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Emofilia: cos’è, diagnosi, sintomi, tipi, terapia e cura
- Aracnodattilia, segno del pollice e del polso, Sindrome di Marfan e di Beals
- Fibrosi cistica polmonare: cos’è, sintomi in neonati e bambini, cure
- Malattia di Huntington: cos’è, ereditarietà, come si trasmette, età di insorgenza
- Anemia falciforme: cosa significa, cause, sintomi e cure
- Talassemia: cos’è, sintomi, cure, differenti tipi ed alimentazione
- Celiachia: cos’è il glutine, in quali alimenti è contenuto ed in quali no?
- Ectrodattilia: cause, cure ed immagini
- Polidattilia; cause, ereditarietà, sindromica e chirurgia
- Sindrome di Beals e padiglione auricolare “accartocciato”: sintomi e cure
- Differenza tra infiammazione cronica granulomatosa e non granulomatosa
- Malattia granulomatosa cronica: causa, trasmissione, sintomi, cura
- Deficit di glucosio-6-fosfato deidrogenasi e favismo: cause, sintomi, terapie
- Quanti cromosomi hanno esseri umani, scimmie, cani, gatti e topi?
- Quanti cromosomi ha chi è affetto da Sindrome di Down?
- Differenza tra paziente asintomatico e portatore sano
- Differenza tra gene e allele
- Cos’è un cromosoma ed a che serve?
- Cos’è un gene ed a che serve?
- Differenza tra malattia genetica, ereditaria e congenita
- Differenza tra genetica ed epigenetica
- Differenza tra genetica e genomica
- Differenza genetica tra uomo e scimmia
- Cosa sono gli alleli ed a che servono?
- Virus e virioni: cosa sono, come sono fatti, come funzionano e come si riproducono
- Riproduzione cellulare e ciclo cellulare
- Meiosi: spiegazione di tutte tappe
- Mitosi: spiegazione delle quattro fasi
- Differenza tra mitocondri e cloroplasti
- Differenza tra citosol e citoplasma
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Organelli (organuli) citoplasmatici della cellula animale: cosa sono ed a che servono?
- Mitocondri: definizione, dimensioni e funzioni
- Citoscheletro: funzioni e struttura
- Ribosomi e reticolo endoplasmatico: cosa sono e che funzioni svolgono?
- Nucleo cellulare: funzioni, dimensioni e membrane nucleari
- Lisosomi: cosa sono? Significato e dimensioni
- Perossisomi: definizione e funzioni
- Membrana plasmatica: definizione e funzioni
- Apparato del Golgi: spiegazione semplice e funzioni
- Citosol: definizione e funzioni
- Differenza tra riproduzione sessuata e asessuata
- Differenza tra vaccini vivi ed attenuati: vantaggi e svantaggi
- Differenza tra immunità specifica ed immunità aspecifica
- Differenza tra linfociti B e T
- Differenza tra immunità umorale e cellulare
- Differenza tra self non self in immunologia
- Differenza tra ciglia e flagelli con esempi
- Differenza tra ciglia e stereociglia: movimento, struttura e funzioni
- Differenza tra ciglia e microvilli: struttura, movimento e funzioni
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
 Si chiama GENOTIPO l’insieme dei geni che compongono il corredo cromosomico di un organismo. Tenendo in considerazione un solo genotipo, ad esempio quello che determina il colore di un fiore, la proteina che darà il colore del fiore deriva dalla combinazione di due geni, uno sul cromosoma paterno e uno materno. I tipi di alleli di quel gene corrispondono al fenotipo per quel carattere (e per carattere in questo caso indichiamo il colore del fiore).
Si chiama GENOTIPO l’insieme dei geni che compongono il corredo cromosomico di un organismo. Tenendo in considerazione un solo genotipo, ad esempio quello che determina il colore di un fiore, la proteina che darà il colore del fiore deriva dalla combinazione di due geni, uno sul cromosoma paterno e uno materno. I tipi di alleli di quel gene corrispondono al fenotipo per quel carattere (e per carattere in questo caso indichiamo il colore del fiore). Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard.
Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard.
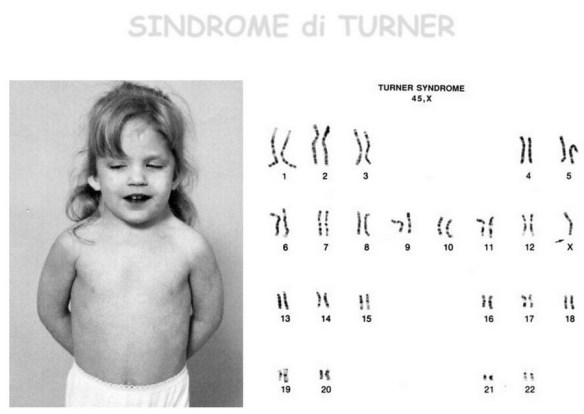 La sindrome di Turner è una sindrome cromosomica caratterizzata da bassa statura, disgenesia gonadica in assenza di ambiguità dei genitali (difetti dello sviluppo dei caratteri sessuali secondari ed infertilità), segni caratteristici del fenotipo esterno ed anomalie di alcuni organi interni.
La sindrome di Turner è una sindrome cromosomica caratterizzata da bassa statura, disgenesia gonadica in assenza di ambiguità dei genitali (difetti dello sviluppo dei caratteri sessuali secondari ed infertilità), segni caratteristici del fenotipo esterno ed anomalie di alcuni organi interni. La schizofrenia è una psicosi cronica caratterizzata dalla persistenza di sintomi di alterazione del pensiero, del comportamento e dell’affettività, da un decorso superiore ai sei mesi, con forte disadattamento della persona ovvero una gravità tale da limitare le normali attività di vita della persona.
La schizofrenia è una psicosi cronica caratterizzata dalla persistenza di sintomi di alterazione del pensiero, del comportamento e dell’affettività, da un decorso superiore ai sei mesi, con forte disadattamento della persona ovvero una gravità tale da limitare le normali attività di vita della persona. Si dice OMOZIGOTE quando un individuo porta due alleli identici per uno stesso gene (AA o aa), mentre ETEROZIGOTE quando un individuo porta due alleli differenti per lo stesso gene (Aa). Il termine omozigote si riferisce quindi ad un gene in cui l’informazione riportata, che determina il fenotipo, dall’allele materno, è identica a quella paterna. Diversamente, negli eterozigoti, il contributo dell’allele materno e paterno è diverso. In tal caso la determinazione fenotipica è correlata ai concetti di dominanza e recessività genetica.
Si dice OMOZIGOTE quando un individuo porta due alleli identici per uno stesso gene (AA o aa), mentre ETEROZIGOTE quando un individuo porta due alleli differenti per lo stesso gene (Aa). Il termine omozigote si riferisce quindi ad un gene in cui l’informazione riportata, che determina il fenotipo, dall’allele materno, è identica a quella paterna. Diversamente, negli eterozigoti, il contributo dell’allele materno e paterno è diverso. In tal caso la determinazione fenotipica è correlata ai concetti di dominanza e recessività genetica. I cromosomi sono la forma in cui si presenta il DNA all’interno della cellula: il lungo filamento di DNA è infatti “impacchettato” fino a formare il cromosoma. Negli eucarioti il DNA è sempre legato a proteine, istoniche e non istoniche, attorno alle quali il filamento di DNA si avvolge a formare complessivamente una struttura chiamata cromatina. La cromatina si può colorare con alcuni coloranti istologici, da cui il nome; se ne possono distinguere due tipi: l’eucromatina, debolmente colorabile, dalla struttura più aperta e quindi trascrizionalmente attiva, e l’eterocromatina, intensamente colorabile, maggiormente condensata (rimane condensata anche in interfase) e trascrizionalmente inattiva. L’eterocromatina può essere ulteriormente distinta in costitutiva e facoltativa. L’eterocromatina costitutiva è costituita da regioni di DNA altamente ripetitivo, costanti in tutte le cellule dell’organismo e nel cromosoma si concentra principalmente a livello del centromero e dei telomeri. L’’eterocromatina facoltativa può diventare condensata e diventare temporaneamente inattiva, inoltre può essere inattivata solo in determinati tessuti o in determinati stadi dello sviluppo.
I cromosomi sono la forma in cui si presenta il DNA all’interno della cellula: il lungo filamento di DNA è infatti “impacchettato” fino a formare il cromosoma. Negli eucarioti il DNA è sempre legato a proteine, istoniche e non istoniche, attorno alle quali il filamento di DNA si avvolge a formare complessivamente una struttura chiamata cromatina. La cromatina si può colorare con alcuni coloranti istologici, da cui il nome; se ne possono distinguere due tipi: l’eucromatina, debolmente colorabile, dalla struttura più aperta e quindi trascrizionalmente attiva, e l’eterocromatina, intensamente colorabile, maggiormente condensata (rimane condensata anche in interfase) e trascrizionalmente inattiva. L’eterocromatina può essere ulteriormente distinta in costitutiva e facoltativa. L’eterocromatina costitutiva è costituita da regioni di DNA altamente ripetitivo, costanti in tutte le cellule dell’organismo e nel cromosoma si concentra principalmente a livello del centromero e dei telomeri. L’’eterocromatina facoltativa può diventare condensata e diventare temporaneamente inattiva, inoltre può essere inattivata solo in determinati tessuti o in determinati stadi dello sviluppo.