
 La sindrome di Prader-Willi (anche chiamata “sindrome di Prader-Labhart-Willi”, in inglese “Prader–Willi syndrome”) è una malattia genetica rara molto eterogenea, sia dal punto di vista clinico sia da quello genetico ed è caratterizzata da anomalie ipotalamico-pituitarie associate a ipotonia grave nel periodo neonatale e nei primi due anni di vita ed alla insorgenza di iperfagia, la quale esita nel rischio di obesità patologica durante l’infanzia e nell’età adulta. La sindrome si associa a difficoltà di apprendimento, disturbi comportamentali e problemi psichiatrici gravi.
La sindrome di Prader-Willi (anche chiamata “sindrome di Prader-Labhart-Willi”, in inglese “Prader–Willi syndrome”) è una malattia genetica rara molto eterogenea, sia dal punto di vista clinico sia da quello genetico ed è caratterizzata da anomalie ipotalamico-pituitarie associate a ipotonia grave nel periodo neonatale e nei primi due anni di vita ed alla insorgenza di iperfagia, la quale esita nel rischio di obesità patologica durante l’infanzia e nell’età adulta. La sindrome si associa a difficoltà di apprendimento, disturbi comportamentali e problemi psichiatrici gravi.
Età di esordio
L’età di esordio della sindrome di Prader-willi è quella neonatale (cioè alla nascita).
Diffusione
La malattia colpisce 1/25.000 nati.
Cause
La malattia ha cause genetiche essendo correlata ad anomalie della regione critica del cromosoma 15 (15q11-q13) ereditata dal padre: è infatti sottoposta a un fenomeno genetico chiamato “imprinting parentale”, per cui sono attivi solo i geni presenti nella regione ereditata dal padre e non dalla madre.
Trasmissione
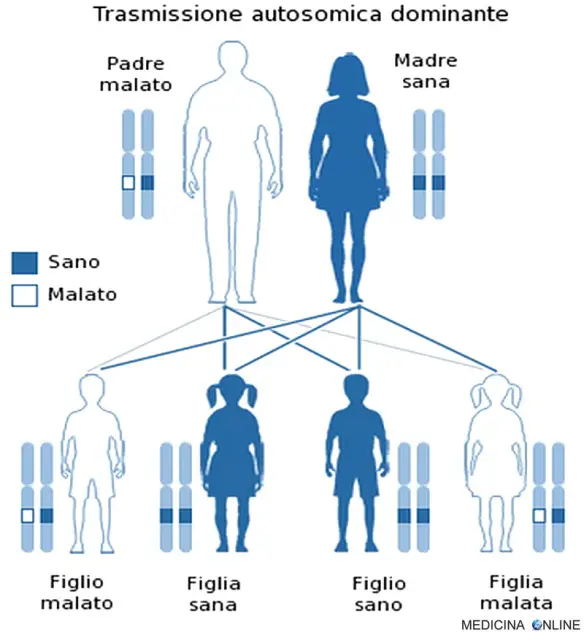
L’anomalia genetica che determina la sindrome di Prader-Willi si trasmette dai genitori ai figli con modalità autosomica dominante, tuttavia la maggior parte dei casi è sporadica e i casi familiari di sindrome di Prader-Willi sono rari. Ad ogni modo ricordiamo al lettore che una malattia è detta a trasmissione autosomica dominante quando basta una singola copia dell’allele difettoso per far sì che la malattia si esprima, a prescindere dal sesso (basta un solo genitore malato). Il figlio di un individuo affetto ha la probabilità del 50% di essere affetto, cioè 1 figlio su 2 è malato e può trasmettere a sua volta la malattia alla metà dei suoi figli (vedi immagine in alto nell’articolo). In questo caso non può esistere un “portatore sano” (cosa che invece si può verificare nella trasmissione autosomica recessiva): chi possiede l’allele alterato, ha la patologia, mentre chi non lo possiede è sano. Di conseguenza da due genitori sani nascono il 100% di figli sani, mentre se entrambi i genitori sono malati allora si avranno il 100% di figli malati. Per approfondire: Differenza tra autosomica dominante e recessiva con esempi
Sintomi e segni
La malattia si manifesta già alla nascita con una grave ipotonia e ritardo nello sviluppo psicomotorio, che comportano problemi alla deglutizione e all’allattamento, ma tende ad attenuarsi parzialmente con l’età. A partire dai due anni di età il bambino mostra invece una costante assenza di sazietà (iperfagia) la quale spinge il bambino a sovralimentarsi: ciò può portare a malnutrizione per eccesso e ad obesità grave. L’obesità è la causa più importante di morbidità e mortalità dei pazienti.
Deficit cognitivi e comportamentali
La malattia è associata a difficoltà di apprendimento e a disturbi comportamentali (comportamento ossessivo-compulsivo oppure manipolativo) e psichiatrici (difficoltà nell’interpretazione e nell’uso appropriato delle informazioni sociali, che comportano disturbi simili a quelli caratteristici dello spettro autistico) di entità variabile. Il deficit cognitivo è estremamente variabile e si associa a difficoltà di apprendimento e a uno sviluppo anomalo del linguaggio, spesso aggravati dai disturbi comportamentali e psicologici.
Caratteristiche facciali e somatiche
Il soggetto ha caratteristiche facciali e somatiche peculiari, come:
- fronte stretta;
- occhi a mandorla;
- labbro superiore sottile;
- bocca rivolta verso il basso;
- mani e piedi molto piccoli;
- scoliosi;
- bassa statura (dovuta a deficit dell’ormone della crescita);
- sviluppo puberale incompleto.
Complicanze
Le complicanze più temibili si verificano in età adulta, dove possono essere presenti:
- obesità molto grave (anche di II o III grado);
- complicanze cardiorespiratorie;
- ipertensione arteriosa;
- maggior rischio di infarto del miocardio ed ictus cerebrale;
- patologie metaboliche;
- complicanze osteoarticolari;
- apnee notturne di tipo ostruttivo, aggravate dalla presenza concomitante di apnee centrali;
- vasculopatie;
- diabete mellito di tipo 2;
- ipercolesterolemia;
- iperglicemia;
- patologia psichiatrica grave;
- idee suicidarie.
Diagnosi
La diagnosi deve basarsi su criteri clinici (criteri di Holm del 1993, rivisti nel 2001) e essere confermata dalle analisi genetiche (tramite test di biologia molecolare utile a verificare le caratteristiche biochimiche della regione 15q11-q13 o tramite indagine citogenetica).
Diagnosi prerenale
Tale sindrome può essere sospettata, in famiglie a rischio, già nell’epoca fetale grazie ad una riduzione dei movimenti fetali, rilevabile all’ecografia. Se una coppia ha già avuto un bambino affetto dalla sindrome, in caso di gravidanze successive è possibile effettuare diagnosi prenatale.
Terapia
Non esiste una terapia specifica risolutiva. La presa in carico deve essere precoce (fin dal periodo neonatale), globale e includere varie figure professionali: ciò può migliorare l’evoluzione della malattia e diminuirne le complicazioni. I trattamenti possibili riguardano in particolare l’ipotonia, i disturbi psichiatrici, i disturbi del comportamento alimentare, i ritardi della crescita (tramite somministrazione dell’ormone della crescita) ed il ritardo della pubertà. Il trattamento con GH (ormone della crescita) può migliorare il tono muscolare, aumentare la velocità di crescita, diminuire il rapporto peso/altezza, diminuire la massa grassa e migliorare la funzionalità respiratoria.
Prognosi
La diagnosi precoce, la terapia multidisciplinare precoce e la terapia con l’ormone della crescita hanno migliorato sensibilmente la qualità della vita di questi bambini. Al momento non esistono dati sugli effetti a lungo termine della terapia con GH negli adulti, in particolare circa l’effetto sui disturbi comportamentali e sul grado di autonomia raggiunto. Negli adulti, le complicazioni legate all’obesità, ai disturbi psichiatrici e all’autonomia rappresentano i problemi più importanti, ma possono essere ridotti tramite dieta specifica, educazione alimentare, psicoterapia e adeguata attività fisica.
Leggi anche:
- Visita neurologica: svolgimento, esami, patologie, quando è necessaria?
- Artrogriposi multipla congenita: cause, sintomi, diagnosi, cure
- Malattie neuromuscolari congenite: contratture congenite dei muscoli e deformità articolari
- Polimiopatie congenite e atrofie muscolari spinali dell’infanzia
- Miopatie endocrine da patologia tiroidea o di altre ghiandole endocrine
- Miopatie metaboliche primitive da accumulo di glicogeno e mitocondriali
- Miopatie secondarie a farmaci, alcolismo e altre sostanze
- Distrofia muscolare in adulti e bambini: sintomi, cause, diagnosi e cure
- Differenze tra la distrofia muscolare di Duchenne e di Becker
- Distrofia muscolare di Duchenne, di Becker e di Emery-Dreifuss
- Distrofia di Landouzy-Dejerine, oftalmoplegia esterna progressiva, sindrome di Kearns-Sayre e distrofia oculofaringea
- Distrofia miotonica di tipo 1 e 2: cause, trasmissione, sintomi, diagnosi, cure
- Distrofia miotonica di Steinert (tipo 1) e distrofia miotonica congenita
- Miopatia miotonica prossimale (malattia di Ricker): cause, sintomi, diagnosi, cure
- Differenza tra distrofia muscolare e sclerosi multipla
- Differenza tra distrofia muscolare e SLA (sclerosi laterale amiotrofica)
- Differenza tra distrofia muscolare ed atrofia muscolare progressiva
- Differenza tra astenia, ipostenia, miastenia, ipotonia, nevrastenia, iperstenia, ipertonia
- Astenia, quando mancano le forze fisiche o mentali: cause, diagnosi, cure
- Ipostenia (miastenia): cause, sintomi, diagnosi, terapie, complicanze, prognosi
- Rabdomiolisi: sintomi, da statine, conseguenze, valori
- Torcicollo spasmodico (distonia cervicale) e spasmi linguali, facciali, oromandibolari, della mano (distonie focali)
- CPK (CK) alto o basso: cause, conseguenze, valori, cure, cosa indica
- Miotonia: cause, sintomi, diagnosi e terapie
- Malattie dei canali del cloro: miotonia congenita di Thomsen
- Malattie dei canali del cloro: miotonia congenita di Becker
- Malattie dei canali del sodio: paramiotonia congenita di Von Eulenburg, paralisi periodica iperkaliemica e normokaliemica
- Malattie dei canali del calcio: paralisi periodica ipokaliemica (malattia di Westphall)
- Miastenia gravis e malattia del timo: cause, sintomi, diagnosi, terapia
- Sindrome miastenica di Lambert-Eaton e altre sindromi miasteniche
- Crampo dello scrivano: cause, sintomi, diagnosi e trattamento
- Distonia mioclonica: cause, sintomi, diagnosi e terapie
- Nevrastenia (esaurimento nervoso): cause, diagnosi, cure
- Ipotonia e atonia: definizione, etimologia, significato, esempi
- Ipotonia muscolare in neonati, adulti, anziani: cause, sintomi, cure, consigli
- Ipotonia nel lattante: cause, sintomi, diagnosi e trattamento
- Iperstenia: definizione, significato, etimologia, fisiologica e patologica
- Ipertonia: definizione, significato, etimologia
- Ipertonia muscolare: cause, tipi, sintomi, diagnosi, terapie
- Cos’è il tono muscolare, a che serve, come mantenerlo?
- Distonia focale e generalizzata: tipi, sintomi, diagnosi e terapie
- Miotonia: cause, sintomi, diagnosi e terapie
- Ipertermia maligna: cause, fattori di rischio, sintomi, rischi, cure, mortalità
- Visita allergologica: svolgimento, esami, preparazione, durata, costo
- Che significa malattia autoimmune? Spiegazione ed esempi
- Discinesia, ipocinesia, ipercinesia, acinesia: significato, esempi
- Esame della sensibilità tattile, dolorifica, termica, vibratoria in neurologia
- Esame delle funzioni motorie e dei riflessi in neurologia
- Esame delle funzioni cerebrali superiori (corticali) in neurologia
- Disturbi della stazione eretta e della deambulazione in neurologia
- Spasmi muscolari e mioclonie: da cosa sono causati?
- Spasmi muscolari e mioclonie: cause, diagnosi e cura delle contrazioni involontarie
- Spasmi muscolari e mioclonie: cura, trattamento e rimedi
- Spasmi muscolari e mioclonie: come si fa la diagnosi?
- Differenza tra tremori, spasmi, miotonia, crampi, fascicolazioni, tic
- Differenza tra discinesia, ipocinesia, ipercinesia, tardiva e primaria
- Sindrome da fatica cronica: cause, sintomi, diagnosi, terapia
- Sindrome dell’uomo rigido: cause, sintomi, diagnosi, terapia
- Sindrome delle gambe senza riposo: cause, sintomi, diagnosi, terapia
- Emiplegia destra, sinistra, spastica, flaccida: significato e riabilitazione
- Emiparesi destra, sinistra, facciale e neonatale: cause, sintomi e cure
- Paraplegia: etimologia, significato, sintomi, cura e riabilitazione
- Tetraplegia: significato, cause, cure e riabilitazione
- Diplegia: definizione, cause e sintomi
- Differenza tra emiplegia, emiparesi, diplegia, paraplegia, tetraplegia
- Differenza emiparesi, diparesi, tetraparesi, monoparesi, triparesi
- Differenza tra paraplegia e diplegia
- Classificazione generale delle paresi e delle plegie
- Stanchezza e debolezza: differenze, cause, rimedi e prevenzione
- Stanchezza pomeridiana tra lavoro e palestra: come ottenere il meglio dal vostro pomeriggio
- Sempre stanco e senza energia al lavoro? Ecco i rimedi
- Sei stanco e di cattivo umore: ecco i cibi che ti danno la carica ed i consigli per avere più energia
- Sei sempre stanco? Ecco le cause meno comuni che non immagineresti mai ed i rimedi
- Sonnolenza e stanchezza cronica: tutte le cause ed i rimedi
- Stress: non basta dormire nel weekend per recuperare
- Ansia da prestazione nello studio e nel lavoro: come superare le tue paure
- Lavori troppo? La tua salute è a rischio: ecco i 10 trucchi per faticare di meno a lavoro
- Combatti lo stress e ritrova il benessere psicofisico con il decalogo del buonumore
- Suicidarsi a causa del Minority Stress: quando appartenere ad una minoranza diventa fonte di discriminazione e sofferenza
- Distrofia muscolare in adulti e bambini: sintomi, cause, diagnosi e cure
- Rabdomiolisi: sintomi, da statine, conseguenze, valori
- Miclono: cause, sintomi, caratteristiche, quando preoccuparsi, cure
- Tic in medicina: significato, tipi, cause, diagnosi, terapie
- Claudicatio intermittens neurologica o vascolare: sintomi, cura, guarigione
- Astenia, sonnolenza, stanchezza: cosa li causa e come curarle
- Asterissi (asterixis) in neurologia: caratteristiche, significato, esecuzione
- Fascicolazioni muscolari, il tremolio spontaneo di un muscolo: cause e cure
- Meralgia parestesica: cause, sintomi, diagnosi, terapie
- Sindromi da compressione nervosa: cause, sintomi, diagnosi e terapie
- Radicolopatia: significato, rimedi casalinghi, attività fisica, integratori, cure
- Sindrome del tunnel carpale: prevenzione, diagnosi e cura di una dolorosa patologia
- Sindrome del tunnel radiale e sindrome del nervo interosseo posteriore
- Sindrome del tunnel cubitale: cause, sintomi, diagnosi e cura
- Sindrome delle fascicolazioni benigne e da crampi: cause, sintomi, cure
- Spasmo in medicina: significato, esempi, cause, diagnosi, cure
- Ernia del disco L5 S1: espulsa, cure naturali, quando operare, guarigione
- Sciatica: quanto dura, come dormire, come farla passare
- Mielopatia: significato, tipi, sintomi, diagnosi, cure, consigli, prognosi
- Mielopatia vascolare: cause, sintomi, diagnosi, cure, rischi
- Mielopatia ischemica e infarto midollare: cause, sintomi, diagnosi, cure
- Tumori vertebro-midollari: tipi, sintomi, diagnosi, terapia, chirurgia, prognosi
- Riflesso di Babinski positivo: sintomi, diagnosi, come evocarlo
- Test di Romberg: cos’è, a che serve, come si esegue
- Segno di Babinski positivo nel neonato e nel bambino: che significa?
- Segno di Babinski nella sclerosi multipla e nella SLA
- Segno di Brudzinski positivo e negativo: semeiotica nella meningite
- Segno di Hoffman positivo in SLA e sclerosi multipla
- Segno di Kernig positivo e negativo: semeiotica nella meningite
- Segno di Lasègue positivo e negativo in semeiotica
- Segno di Wasserman (Lasègue inverso) positivo in semeiotica
- Segno di Binda: cos’è e come si esegue
- Segno di Amoss (o del tripode): cos’è e come si esegue
- Segno di Magnus-De Klein: cos’è e come si esegue
- Segno di Adson: significato, esecuzione, patologie indicate
- Segni meningei e irritazione meningi in bambini ed adulti
- Esame vestibolare positivo o negativo: come si svolge, è fastidioso, a cosa serve?
- Analisi posturale: un esame che individua gli squilibri corporei
- Esame audiometrico: procedura, valori normali e patologici, costo
- Esame impedenzometrico: cos’è, come si svolge, risultati, valori normali
- Tic, ritmie, movimenti stereotipati, acatisia e trasalimento nel paziente neurologico
- Esame della motilità oculare e disturbi dei movimenti coniugati in neurologia
- Sordità, esame dell’udito e tecniche audiologiche speciali in neurologia
- Visita otorinolaringoiatrica: come si svolge, fa male, costo
- Esame della vista (misurazione della acuità visiva): come si svolge, a che serve
- Visita oculistica: in cosa consiste, come si svolge, dove farla, costo
- Esame del campo visivo: risultati, costo, quanto dura, interpretazione
- Differenze tra neurologo e psichiatra
- Differente approccio di psicologo, psicoterapeuta e psichiatra
- Differenze tra le varie scuole di psicoterapia: quale la più efficace?
- Psiconeuroendocrinoimmunologia
- Test di Rorschach: immagini, a cosa serve, interpretazione
- Morbo di Parkinson: cause, sintomi, decorso, terapie
- Morbo di Alzheimer: cause, sintomi, decorso, terapie
- Differenza tra morbo di Alzheimer, demenza senile, vascolare e reversibile
- Sclerosi laterale amiotrofica (SLA): cause, sintomi, diagnosi e prognosi
- Sclerosi multipla: cause, sintomi, diagnosi e prognosi
- Differenze tra sclerosi laterale amiotrofica e sclerosi multipla
- Demenza senile: cause, sintomi, decorso e cure
- Demenza da corpi di Lewy: cause, decorso, Parkinson, aspettativa di vita
- Atrofia muscolare progressiva: cause, sintomi, cura, aspettativa di vita
- Atrofia muscolare spinale: sintomi, trasmissione, tipi e cure
- Differenze tra atrofia muscolare progressiva e sclerosi laterale amiotrofica
- Parestesie: significato, cause, rischi, diagnosi, cure, rimedi, esercizi
- Epilessia infantile ed in adulti: cause, sintomi, diagnosi, cosa fare
- Epilessia: come riconoscere un attacco e soccorrere un ammalato
- Differenza tra epilessia e convulsioni
- Differenza tra epilessia e sincope
- Differenza tra epilessia parziale e generalizzata
- Epilessia: riconoscere in tempo l’arrivo di una crisi e come comportarsi
- Epilessia infantile: come comportarsi col proprio figlio?
- Non riconoscere i volti dei propri cari: la prosopagnosia, cause, test e cure
- Spina bifida e difetti di chiusura del tubo neurale nel feto: trasmissione, prevenzione, diagnosi e cura
- Mielomeningocele, spina bifida e schisi vertebrale: complicanze, cura, prevenzione
- Encefalocele: cause, sintomi, diagnosi, cura e prevenzione
- Mielopatia: significato, tipi, sintomi, diagnosi,cure, consigli, prognosi
- Anche gli esseri umani possono avere una coda
- Transilluminazione della testa di un neonato per la diagnosi di idrocefalo
- Idrocefalo: cause, terapia, conseguenze, aspettativa di vita
- Idrocefalo nel feto e neonatale: conseguenze e cura
- Pressione intracranica e pressione di perfusione cerebrale
- Meningismo: triade, segni, cause, diagnosi, definizione e cura
- Sindrome di Arnold-Chiari: linguaggio, aspettative di vita, invalidità, mortalità
- Siringomielia (malattia di Morvan): cause, sintomi, diagnosi, cure
- Siringobulbia: cause, sintomi, diagnosi e trattamento
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Meningite: contagio, sintomi, vaccino, gravità e profilassi
- Shunt cerebrale e intervento per il drenaggio permanente
- Macrocefalo in neonato e bambino: sintomi, cure e ritardo psicomotorio
- Pressione intracranica e pressione di perfusione cerebrale
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Ventricoli cerebrali: anatomia e funzioni in sintesi
- Segni meningei e irritazione meningi in bambini ed adulti
- Meningite: incubazione, fulminante, sintomi, contagio e cura
- Encefalite: conseguenze, è contagiosa, danni, si guarisce?
- Mielite: infettiva, cervicale, dorsale, trasversa, si guarisce?
- Emorragia subaracnoidea: cause, conseguenze, linee guida
- Ematoma subdurale: cos’è, da quale malattia è provocata, recidivo, decorso
- Glasgow Coma Scale per la classificazione del coma
- Pediatric Glasgow Coma Scale in italiano: scala pediatrica del coma
- Differenza tra meningite e meningismo: qual è più grave?
- Differenza tra meningite, encefalite, meningoencefalite, encefalomielite
- Differenza tra meningite virale e batterica
- Puntura lombare: complicanze, risultati, è dolorosa, a che serve?
- Meningi: anatomia, funzioni e patologia in sintesi
- Liquido cefalorachidiano: dove si trova, perdita dal naso, prelievo
- Differenza idrocefalo iperteso, normoteso, comunicante, ostruttivo
- Differenza tra virus e batteri: chi è più pericoloso? Diagnosi, sintomi e terapia
- Differenza tra toracentesi, paracentesi e rachicentesi
- Narcolessia: cause, sintomi, cure e terapia farmacologica
- Catatonia: significato, definizione, cause, sinonimi e cure
- Cataplessia: causa, significato, nel sonno, cura ed etimologia
- Catalessia in medicina: cause, sintomi, nel sonno e cure
- Differenza tra catatonia, catalessia e cataplessia
- Paralisi del sonno e allucinazioni ipnagogiche: cause, pericoli, rimedi
- Che cos’è un attacco ischemico transitorio (TIA)? Impara a riconoscerlo e potrai salvare una vita, anche la tua
- Aneurisma cerebrale rotto e non rotto: cause, sintomi, diagnosi e cura
- Ictus, emorragia cerebrale cerebrale e TIA: cosa fare e cosa assolutamente NON fare
- Emorragia cerebrale da caduta e trauma cranico: sintomi, diagnosi e cure
- Emorragia cerebrale: non operabile, coma, morte, si può guarire?
- Emorragia cerebrale: operazione e tempi di riassorbimento
- Coma da emorragia cerebrale: quanto può durare?
- Differenza tra stato vegetativo, di minima coscienza, coma, sonno e stato soporoso
- Morte cerebrale: diagnosi, sintomi, risveglio, durata, si può guarire?
- Coma: cause, risveglio, tipi, quanto dura, fasi, segni, irreversibile
- Differenza tra coma e coma farmacologico
- Elettroencefalogramma: preparazione, alterazioni, costo, rischi
- Sindrome locked-in: cause, riabilitazione, respirazione, cure
- Stato di minima coscienza: evoluzione, risveglio, riabilitazione
- Stato vegetativo: risveglio, riabilitazione, durata e caratteristiche
- Commozione cerebrale: cos’è, cosa fare, conseguenze, tempi di recupero
- Differenza tra commozione cerebrale, trauma cranico e contusione cerebrale
- Trauma cranico: ematoma, commotivo, sintomi tardivi, cosa fare
- Differenza tra sindrome locked-in e stato vegetativo
- Differenza tra stato vegetativo persistente e permanente
- Differenza tra stato vegetativo e stato di minima coscienza
- Emorragia cerebrale: cause, sintomi premonitori, diagnosi e cura
- Differenza tra morte clinica, biologica, legale, apparente, improvvisa ed istantanea
- Poligono di Willis: anatomia e varianti anatomiche
- Anosognosia e Sindrome neglect: significato, test e trattamento
- Sindrome neglect (negligenza spaziale unilaterale): cura e riabilitazione
- Sistema nervoso: com’è fatto, a che serve e come funziona
- Sistema nervoso simpatico: funzioni
- Sistema nervoso parasimpatico: funzioni
- Differenza tra afasia, disartria ed aprassia
- Area di Broca: funzioni ed afasia di Broca
- Area di Wernicke: funzioni ed afasia di Wernicke
- Differenza tra afasia di Broca e di Wernicke
- Cervelletto: anatomia esterna ed interna
- Cervelletto: le lesioni cerebellari più comuni
- Le funzioni del cervelletto: apprendimento e correzione dei movimenti del corpo
- Com’è fatto il cervello, a che serve e come funziona la memoria?
- Cervello maschile e femminile: quali sono le differenze?
- Sistema nervoso autonomo simpatico e parasimpatico: anatomia e funzioni
- Ipotalamo: anatomia, struttura e funzioni
- Differenze tra ipotalamo, ipofisi, neuroipofisi e adenoipofisi
- Differenza tra midollo osseo e spinale
- Differenza tra sistema nervoso centrale e periferico: anatomia e funzioni in sintesi
- A cosa serve il midollo osseo?
- Differenza tra midollo osseo e cellule staminali
- Differenza tra midollo spinale e allungato
- Differenza tra epifisi, diafisi, metafisi ed ipofisi
- Patologie di ipotalamo e ipofisi
- Ipofisi (ghiandola pituitaria): anatomia, funzioni e ormoni secreti
- Asse ipotalamo-ipofisario: fisiologia e ormoni rilasciati
- Si può morire di epilessia?
- Tumore al cervello: operato mentre suona la chitarra e canta Yesterday
- Encefalopatia epatica: terapia, morte, sopravvivenza, dieta, prognosi
- Ammoniemia (ammoniaca nel sangue): valori e dieta per iperammoniemia
- Esami per valutare funzionalità renale ed insufficienza renale
- Differenza tra insufficienza renale acuta, cronica e dialisi
- Encefalopatia uremica nell’uremia: sintomi e alito di urina
- Coma uremico e uremia: sintomi e morte del paziente
- Ipercapnia: valori, terapia, conseguenze e trattamento
- I muscoli: come sono fatti, come funzionano e cosa rischiano quando ti alleni
- Differenza tra muscoli adduttori e abduttori
- Differenza tra muscoli agonisti, antagonisti e sinergici
- Muscoli respiratori volontari ed involontari
Lo Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram o su Pinterest, grazie!
