 Sappiamo che le affinità fra gemelli omozigoti sono molte: condividono in fondo lo stesso identico patrimonio genetico, e se crescono assieme spesso si influenzano vicendevolmente per quanto riguarda il comportamento. In alcuni casi hanno dei gusti marcatamente differenti; quasi sempre però mostrano una comprensione reciproca che, vista dall’esterno, può apparire straordinaria. Ma la storia delle gemelle Gibbons contiene un elemento più viscerale, inspiegabile, come se questa sintonia fosse arrivata ad un livello superiore e ancora oggi impossibile da spiegare.
Sappiamo che le affinità fra gemelli omozigoti sono molte: condividono in fondo lo stesso identico patrimonio genetico, e se crescono assieme spesso si influenzano vicendevolmente per quanto riguarda il comportamento. In alcuni casi hanno dei gusti marcatamente differenti; quasi sempre però mostrano una comprensione reciproca che, vista dall’esterno, può apparire straordinaria. Ma la storia delle gemelle Gibbons contiene un elemento più viscerale, inspiegabile, come se questa sintonia fosse arrivata ad un livello superiore e ancora oggi impossibile da spiegare.
June e Jennifer Gibbons erano nate nell’isola Barbados l’11 aprile del 1963. I genitori si trasferirono ad Haverfordwest, nel Galles (il padre era luogotenente nella RAF) poco dopo la nascita delle gemelline. Si trattava dell’unica famiglia di colore della città, e certamente il problema dell’integrazione deve aver pesato sullo sviluppo delle piccole gemelle. Divenne presto evidente che le bambine avevano alcune difficoltà di linguaggio, tanto che soltanto la mamma Gloria era in grado di capire quello che farfugliavano, e in alcuni casi nemmeno lei. A causa di questi disordini linguistici, June e Jennifer crebbero senza legare con gli altri bambini, sempre sole e chiuse nel loro mondo.
La scuola, come è comprensibile, fu per loro un trauma severo: rifiutavano di parlare, scrivere o leggere, e gli insegnanti cominciarono a mandarle a casa prima della fine dell’orario per dare loro qualche minuto di vantaggio sui bulli che le molestavano in continuazione. Fu in quel periodo che cominciarono ad essere chiamate the silent twins (gemelle silenziose).
Se avevano eretto un muro impenetrabile per il mondo, all’interno del loro “spazio protetto” vivevano però una realtà differente.
Le gemelle avevano sviluppato un loro linguaggio, incomprensibile agli estranei (criptofasia), e dei giochi segreti particolari e complicati. Giocavano a “specchiarsi” l’una nell’altra, imitando a vicenda le azioni compiute; la sera decidevano chi delle due, al risveglio mattutino, avrebbe respirato per prima, e finché questo respiro non veniva avvertito l’altra sorella doveva giacere immobile, come morta. Mentre in classe non c’era verso di costringerle a leggere, nella serenità della loro cameretta erano avide divoratrici di libri, e riempivano i loro quaderni di racconti, disegni e romanzi scritti a quattro mani. Le pagine erano riempite di caratteri minuscoli, tanto che fra una riga blu e l’altra dei fogli dei loro diari trovavano spazio quattro righe di testo.
All’età di 14 anni, avevano ormai escluso dalla loro vita praticamente chiunque: compagni, conoscenti, mamma, papà, e i due fratelli. Soltanto alla piccola Rosie, la sorella minore, era consentito entrare sporadicamente nel loro universo. Ormai il silenzio era divenuto un voto vero e proprio, e il linguaggio segreto utilizzato fra di loro era sempre più impenetrabile. Si muovevano con gesti lenti, all’unisono, senza dubbio rispettando le regole di un oscuro gioco. I diversi psicologi e terapeuti non riuscirono a fare nulla per renderle più sociali, e le ragazze sprofondarono inesorabilmente nel loro rapporto esclusivo.
Le gemelle, a onor del vero, provarono ad uscire dall’isolamento attraverso la scrittura. Due dei loro romanzi, Pepsi-Cola Addict e Discomania, firmati rispettivamente da June e Jennifer, vennero pubblicati a loro spese, senza però attrarre l’attenzione sperata. Qualche breve flirt con dei ragazzi americani non portò ugualmente a nulla di importante. Il problema vero sorse quando le due cominciarono ad avere dei comportamenti delinquenziali: piccoli crimini, che culminarono però in due episodi di incendi dolosi, appiccati dalle gemelle alle scuole speciali che frequentavano. Il loro rapporto di amore si mischiava inoltre ad accessi di odio violento, visto che un giorno Jennifer aveva tentato di strangolare June con un cavo della radio, e una settimana dopo June aveva spinto Jennifer giù da un ponte nel fiume sottostante.
La risposta del sistema giudiziario fu particolarmente dura: reputate pericolose, le due ragazze vennero rinchiuse nel Broadmoor Hospital, un ospedale di massima sicurezza per malati mentali. Lì, insieme a maniaci, psicopatici e schizofrenici, passarono 14 anni della loro vita, incontrandosi soltanto in orari precisi.
Le gemelle avevano fin dall’inizio pattuito che se una di loro fosse morta, l’altra avrebbe rotto il patto del silenzio, avrebbe cominciato a parlare, e vissuto una vita normale. Nel corso degli anni di reclusione forzata, erano arrivate alla drammatica conclusione che fosse necessario che una delle due morisse: non sarebbero mai state libere, se non tramite il sacrificio.
La loro biografa ed amica, Marjorie Wallace, raccontò il momento in cui le rivelarono il loro piano:
Portai mia figlia – credo avesse circa otto anni – a prendere il tè con le gemelle. Dovevamo passare per tutte queste porte chiuse a chiave, fino alla grande sala. Jennifer e June erano là, sempre piuttosto allegre, e ci portavano il tè su un vassoio con piccoli biscotti. Ci sedemmo e cominciammo a chiacchierare. Di colpo, nel bel mezzo della conversazione, Jennifer disse: ” Marjorie-Marjorie-Marjorie, io morirò”. Io dissi: “Non essere stupida, Jennifer. Sei in buona salute”. Mi guardò e mi disse: “Abbiamo deciso, io morirò”. […] Poi June disse: “Sì, abbiamo deciso”. Mi passarono dei biglietti, e c’era scritto che la decisione era che Jennifer sarebbe dovuta morire per liberare June. June era nata per prima, June aveva più talento, June era più estroversa. Aveva il diritto di vivere. June poteva vivere per entrambe, e Jennifer no.
Il 9 marzo 1993 le due sorelle (che allora avevano 29 anni) vennero spostate da Broadmoor alla Caswell Clinic a Bridgend, dove sarebbero state sottoposte finalmente a un regime più libero. Nel minibus che le trasportava, però, Jennifer di colpo poggiò la testa sulla spalla della sorella. June dichiarerà in seguito: “Pensavo fosse stanca. Sembrava che dormisse, ma i suoi occhi erano aperti e sbarrati”. Quando il bus arrivò alla clinica, non si riuscì a svegliare Jennifer. Portata all’ospedale, vi morì poche ore dopo.
L’autopsia rivelò che Jennifer era morta di miocardite acuta, un’infiammazione del muscolo cardiaco. Secondo gli anatomopatologi, potevano esserci circa 40 motivi diversi per una tale infiammazione; eppure il Dr. Knight, patologo, disse di non avere mai visto un cuore così severamente infiammato senza alcuna ragione evidente.
Rimasta sola, June fu effettivamente in grado di conquistarsi una vita più comune, senza farmaci o cliniche. Ancora oggi rifugge dai riflettori dei media; conduce una vita serena e anonima aiutando di tanto in tanto i vecchi genitori, accettata finalmente dalla comunità, senza grossi problemi, e tenta di lasciarsi il passato dietro le spalle.
La morte di Jennifer resta un mistero per la medicina.
Leggi anche:
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
Condividi questo articolo:

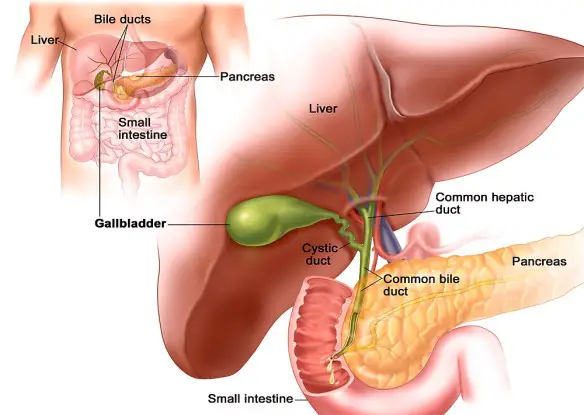 La cirrosi epatica è l’espressione di una tappa terminale di danno cronico del fegato indotto da fattori lesivi di diversa origine. Al danno cronico il fegato ripara compensando attraverso la formazione di “cicatrici” costituite da tessuto fibroso e attraverso la rigenerazione delle cellule perdute. L’espressione esasperata di tali processi porta ad una estesa fibrosi e ai noduli di rigenerazione; a lungo andare ne consegue una progressiva riduzione della massa funzionante epatica (causa di insufficienza dell’organo nelle fasi di malattia avanzata), fenomeni infiammatori e necrotici ed un sovvertimento della architettura vascolare intraepatica con conseguente ipertensione portale e cirrosi conclamata.
La cirrosi epatica è l’espressione di una tappa terminale di danno cronico del fegato indotto da fattori lesivi di diversa origine. Al danno cronico il fegato ripara compensando attraverso la formazione di “cicatrici” costituite da tessuto fibroso e attraverso la rigenerazione delle cellule perdute. L’espressione esasperata di tali processi porta ad una estesa fibrosi e ai noduli di rigenerazione; a lungo andare ne consegue una progressiva riduzione della massa funzionante epatica (causa di insufficienza dell’organo nelle fasi di malattia avanzata), fenomeni infiammatori e necrotici ed un sovvertimento della architettura vascolare intraepatica con conseguente ipertensione portale e cirrosi conclamata. Se dovete trovare una strada o l’abitazione di un amico è meglio chiedere informazioni e orientarvi, seguendo le indicazioni presenti sui vari cartelloni. Per quanto possiate perdervi, in questa maniera, almeno non rischiate di spegnere il vostro cervello. Questi ha a disposizione delle zone atte all’orientamento. Parti che rischiano di essere danneggiate dall’uso costante di GPS e navigatore. A dimostrare tutto ciò uno studio pubblicato su Nature Communications. Sono stati osservati alcuni automobilisti mentre camminavano per uno dei quartieri più affollati di Londra, ossia Soho. Sono state effettuate vere e proprie scansioni cerebrali. In questa maniera sono state osservate le reazioni interne all’Ippocampo e alla Corteccia Prefrontale.
Se dovete trovare una strada o l’abitazione di un amico è meglio chiedere informazioni e orientarvi, seguendo le indicazioni presenti sui vari cartelloni. Per quanto possiate perdervi, in questa maniera, almeno non rischiate di spegnere il vostro cervello. Questi ha a disposizione delle zone atte all’orientamento. Parti che rischiano di essere danneggiate dall’uso costante di GPS e navigatore. A dimostrare tutto ciò uno studio pubblicato su Nature Communications. Sono stati osservati alcuni automobilisti mentre camminavano per uno dei quartieri più affollati di Londra, ossia Soho. Sono state effettuate vere e proprie scansioni cerebrali. In questa maniera sono state osservate le reazioni interne all’Ippocampo e alla Corteccia Prefrontale.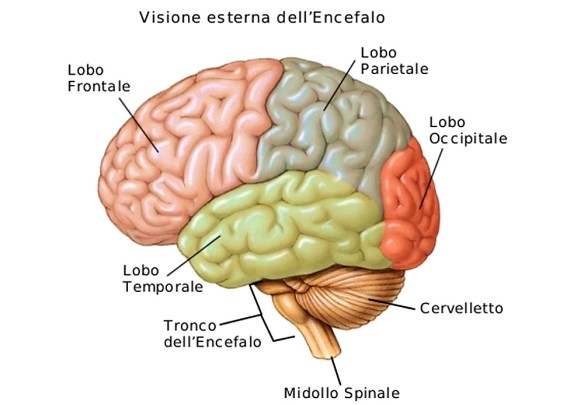 La malattia (o morbo o còrea) di Huntington è una patologia ereditaria causata dalla degenerazione di neuroni situati in specifiche aree cerebrali – striato e corteccia cerebrale – e caratterizzata da una generale atrofia del cervello. I sintomi iniziali possono essere bruschi mutamenti dell’umore, apatia, irritabilità, depressione e rabbia, difficoltà nella guida, nell’imparare cose nuove o nel prendere una decisione. Altri possono presentare cambiamenti nella scrittura e movimenti involontari delle dita, dei piedi, del viso o del tronco (chiamati “còrea” dal termine greco che significa “danza”). In altri casi possono insorgere disturbi dell’equilibrio e del coordinamento motorio con accentuato rischio di cadute. L’ordine di comparsa di questi sintomi e la gravità possono variare notevolmente da un individuo all’altro, così come l’età d’insorgenza.
La malattia (o morbo o còrea) di Huntington è una patologia ereditaria causata dalla degenerazione di neuroni situati in specifiche aree cerebrali – striato e corteccia cerebrale – e caratterizzata da una generale atrofia del cervello. I sintomi iniziali possono essere bruschi mutamenti dell’umore, apatia, irritabilità, depressione e rabbia, difficoltà nella guida, nell’imparare cose nuove o nel prendere una decisione. Altri possono presentare cambiamenti nella scrittura e movimenti involontari delle dita, dei piedi, del viso o del tronco (chiamati “còrea” dal termine greco che significa “danza”). In altri casi possono insorgere disturbi dell’equilibrio e del coordinamento motorio con accentuato rischio di cadute. L’ordine di comparsa di questi sintomi e la gravità possono variare notevolmente da un individuo all’altro, così come l’età d’insorgenza.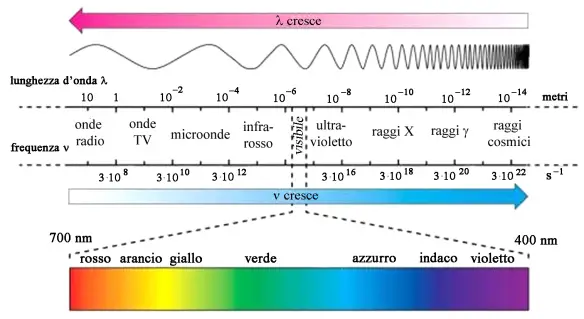 In fisica lo spettro elettromagnetico indica l’insieme di tutte le possibili frequenze delle radiazioni elettromagnetiche. Pur essendo lo spettro continuo, è possibile una suddivisione puramente convenzionale ed indicativa in vari intervalli o bande di frequenza, dettata a partire dallo spettro ottico. L’intero spettro è suddiviso nella parte di spettro visibile che dà vita alla luce e le parti di spettro non visibile a lunghezza d’onda maggiori e minori dello spettro visibile. Le onde di maggiore lunghezza d’onda dal visibile alle onde radio hanno poca energia e risultano scarsamente dannose, le radiazioni comprese tra l’ultravioletto ed i raggi gamma invece hanno più energia, sono ionizzanti e quindi possono danneggiare gli esseri viventi. Dalla parte dello spettro, dove la luce ha lunghezza d’onda maggiore, cioè oltre il rosso, si trova la zona denominata infrarossa. Quest’ultima va da 0,7 µm a 0,4 mm. Quindi, viene la zona delle microonde, con lunghezze d’onda da 0,4 mm a 100 cm. Oltre a questa, vi sono tre campi di onde radio: onde corte da 1 m a 100 m; onde medie da 200 m a 600 m; onde lunghe superiori a 600 m. Le onde radio possono essere generate da scariche che producono onde elettromagnetiche.
In fisica lo spettro elettromagnetico indica l’insieme di tutte le possibili frequenze delle radiazioni elettromagnetiche. Pur essendo lo spettro continuo, è possibile una suddivisione puramente convenzionale ed indicativa in vari intervalli o bande di frequenza, dettata a partire dallo spettro ottico. L’intero spettro è suddiviso nella parte di spettro visibile che dà vita alla luce e le parti di spettro non visibile a lunghezza d’onda maggiori e minori dello spettro visibile. Le onde di maggiore lunghezza d’onda dal visibile alle onde radio hanno poca energia e risultano scarsamente dannose, le radiazioni comprese tra l’ultravioletto ed i raggi gamma invece hanno più energia, sono ionizzanti e quindi possono danneggiare gli esseri viventi. Dalla parte dello spettro, dove la luce ha lunghezza d’onda maggiore, cioè oltre il rosso, si trova la zona denominata infrarossa. Quest’ultima va da 0,7 µm a 0,4 mm. Quindi, viene la zona delle microonde, con lunghezze d’onda da 0,4 mm a 100 cm. Oltre a questa, vi sono tre campi di onde radio: onde corte da 1 m a 100 m; onde medie da 200 m a 600 m; onde lunghe superiori a 600 m. Le onde radio possono essere generate da scariche che producono onde elettromagnetiche. m; la banda compresa tra 0,25 e 0,31
m; la banda compresa tra 0,25 e 0,31 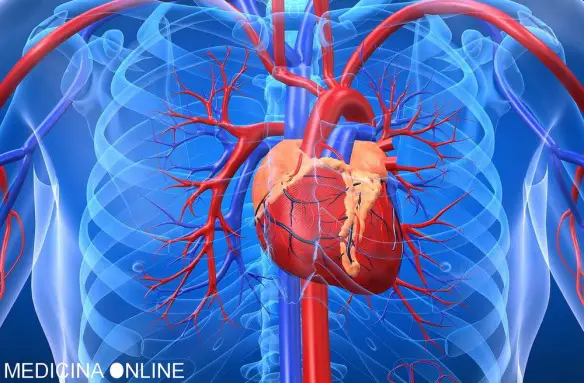 Con “ischemia” si intende una diminuzione – parziale o totale – dell’apporto di sangue a una regione di un organo o a un tessuto causata da un problema cardio-circolatorio di varia natura. Conseguentemente a questo fenomeno, le cellule non ricevono l’ossigeno nella quantità necessaria, provocando diversi danni la cui entità dipende, ad esempio, dall’area del corpo colpita e dalla durata dell’ischemia. Gli organi più sensibili in tal senso sono ad esempio il cuore e l’encefalo: nel primo caso si può assistere a un’ischemia del miocardio, il tessuto muscolare del cuore, nel secondo caso si può verificare un ictus ischemico. Maggiore è il tempo durante il quale il tessuto è colpito da tale fenomeno, minore sarà la possibilità di recupero: per questo è fondamentale riuscire a riconoscere tempestivamente i sintomi di un’ischemia per poter intervenire il prima possibile. A livello cellulare, l’effetto che si manifesta è il rallentamento o l’interruzione della catena respiratoria, con il conseguente accumulo di sostanze quali l’acido lattico (per attivazione della glicolisi anaerobia) fino ad arrivare al disequilibrio della distribuzione ionica e necrosi cellulare. Tra le cause più frequenti di ischemia si ricordano l’aterosclerosi, l’embolia e la vasocostrizione prolungata.
Con “ischemia” si intende una diminuzione – parziale o totale – dell’apporto di sangue a una regione di un organo o a un tessuto causata da un problema cardio-circolatorio di varia natura. Conseguentemente a questo fenomeno, le cellule non ricevono l’ossigeno nella quantità necessaria, provocando diversi danni la cui entità dipende, ad esempio, dall’area del corpo colpita e dalla durata dell’ischemia. Gli organi più sensibili in tal senso sono ad esempio il cuore e l’encefalo: nel primo caso si può assistere a un’ischemia del miocardio, il tessuto muscolare del cuore, nel secondo caso si può verificare un ictus ischemico. Maggiore è il tempo durante il quale il tessuto è colpito da tale fenomeno, minore sarà la possibilità di recupero: per questo è fondamentale riuscire a riconoscere tempestivamente i sintomi di un’ischemia per poter intervenire il prima possibile. A livello cellulare, l’effetto che si manifesta è il rallentamento o l’interruzione della catena respiratoria, con il conseguente accumulo di sostanze quali l’acido lattico (per attivazione della glicolisi anaerobia) fino ad arrivare al disequilibrio della distribuzione ionica e necrosi cellulare. Tra le cause più frequenti di ischemia si ricordano l’aterosclerosi, l’embolia e la vasocostrizione prolungata.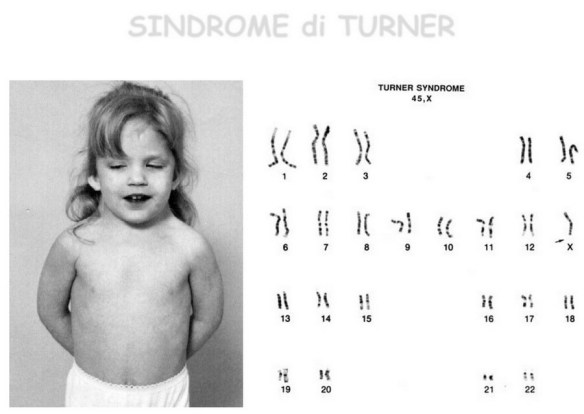 La sindrome di Turner è una sindrome cromosomica caratterizzata da bassa statura, disgenesia gonadica in assenza di ambiguità dei genitali (difetti dello sviluppo dei caratteri sessuali secondari ed infertilità), segni caratteristici del fenotipo esterno ed anomalie di alcuni organi interni.
La sindrome di Turner è una sindrome cromosomica caratterizzata da bassa statura, disgenesia gonadica in assenza di ambiguità dei genitali (difetti dello sviluppo dei caratteri sessuali secondari ed infertilità), segni caratteristici del fenotipo esterno ed anomalie di alcuni organi interni. L’ustione è una lesione dei tessuti tegumentari (pelle ed annessi cutanei) provocata dall’azione di calore, sostanze chimiche, corrente elettrica o radiazioni. Possono essere di varia entità secondo l’intensità della temperatura, la durata del contatto e lo stato fisico della sostanza ustionante (solida, liquida o gassosa); in relazione alla gravità vengono distinte in primo, secondo,terzo e quarto grado.
L’ustione è una lesione dei tessuti tegumentari (pelle ed annessi cutanei) provocata dall’azione di calore, sostanze chimiche, corrente elettrica o radiazioni. Possono essere di varia entità secondo l’intensità della temperatura, la durata del contatto e lo stato fisico della sostanza ustionante (solida, liquida o gassosa); in relazione alla gravità vengono distinte in primo, secondo,terzo e quarto grado.