
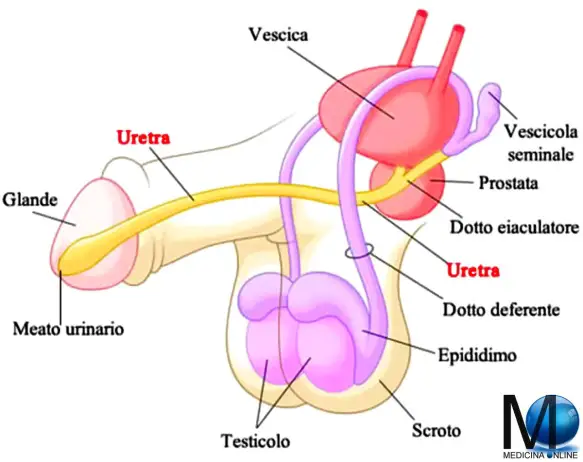
 Con “vescicolite” si intende un’infiammazione – spesso causata da infezione – delle vescicole seminali, due strutture che fanno parte dell’apparato riproduttore maschile, poste vicino alla parete posteriore della prostata. Clinicamente possiamo distinguere forme acute e croniche.
Con “vescicolite” si intende un’infiammazione – spesso causata da infezione – delle vescicole seminali, due strutture che fanno parte dell’apparato riproduttore maschile, poste vicino alla parete posteriore della prostata. Clinicamente possiamo distinguere forme acute e croniche.
Nelle forme acute, sovente insorgenti nel corso di una gonorrea trascurata, si ha il quadro classico di una prostatite acuta: dolore perineale, inguinale, ipogastrico; tenesmo rettale; minzione frequente e dolorosa; febbre; compromissione dello stato generale. All’esplorazione rettale si evidenziano vescicole grosse e molto dolorabili. In rari casi l’ascesso vescicolare può esitare in una peritonite.
Le forme croniche possono rimanere latenti, determinare una sintomatologia soggettiva molto sfumata, oppure generare importanti alterazioni funzionali. Nessun sintomo è patognomonico di vescicolite cronica, pertanto spesso i pazienti sono inquadrati in diagnosi non esatte come prostatiti, cistiti o uretriti.
Il dolore, d’intensità variabile, è localizzato al perineo ed irradiato diversamente da caso a caso (verso il funicolo spermatico ed il testicolo, verso l’uretra, verso la radice della coscia, meno frequentemente verso l’ipogastrio, la fossa iliaca e l’articolazione sacroiliaca).
I disturbi urinari insorgono quando alla vescicolite si associ una uretrite posteriore o una prostatite, per cui si avrà pollachiuria e stranguria.
I disturbi genitali spesso costituiscono la causa della consultazione del medico. Questi possono consistere in turbe della sfera sessuale (frequenti e dolorose erezioni notturne; eiaculazione precoce; erezione difficile, incompleta; impotenza; emospermia) e in turbe della fertilità con quadri seminali caratterizzati da:
- oligoastenospermia
- necrospermia
- teratospermia
- agglutinazione degli spermatozoi
- aumento della viscosità
- aumento del pH
- crescita uniforme di oltre 1000 batteri patogeni per ml o di oltre10.000 batteri non patogeni nel liquido seminale diluito 1:2
- leucospermia (oltre 1 milione di GB x ml)
E’ stata supposta un’azione diretta degli agenti batterici e dei leucociti sugli spermatozoi; tuttavia l’impatto delle infezioni delle vie seminali e delle ghiandole accessorie sull’infertilità maschile resta ancora oggetto di controversia.
Nelle forme che evolvono in marcata atrofia fibrosa si avrà infine la contrazione progressiva del volume dell’eiaculato (oligoposia secondaria).
Leggi anche:
- Vescicola seminale: posizione, anatomia e funzioni in sintesi
- Com’è fatto il pene al suo interno?
- Composizione, caratteristiche e produzione dello sperma
- Come avere la più potente erezione della tua vita senza farmaci
- Esercizi per allungamento del pene (massaggio Jelqing): funzionano? Tecnica e rischi
- Anorgasmia: quando manca l’orgasmo, cause e rimedi
- Erezione debole o assente da cause psicologiche: cura e rimedi
- Scroto: dimensioni, anatomia e funzioni in sintesi
- Testicoli e scroto: dimensioni, anatomia e funzioni in sintesi
- L’autopalpazione del testicolo ti salva dal cancro testicolare
- Torsione del testicolo: sintomi, cure, conseguenze, neonati. E’ doloroso?
- Impianto di protesi testicolare: quando, come e perché si effettua
- Cancro del testicolo: prevenzione, diagnosi, stadiazione, cure
- Quante volte al giorno è normale urinare? Vescica iperattiva e ansia
- Apparato urinario: anatomia e fisiologia [SCHEMA]
- Differenza tra tumore benigno, maligno, neoplasia, cancro e metastasi
- Eiaculazione retrograda: quando lo sperma non esce o è troppo poco
- Differenza tra poliuria e polidipsia
- Differenza tra anuria ed oliguria
- Differenza tra anuria e ritenzione urinaria
- Differenza tra poliuria e pollachiuria
- Differenza tra esame delle urine ed urinocoltura
- Perché viene la cistite e come curarla?
- Si può vivere senza reni? Conseguenze della nefrectomia
- Frequenza defecazione: quante volte al giorno è normale andare di corpo?
- Fa male trattenere l’urina troppo a lungo? Per quale motivo?
- Idratazione corretta: quanta acqua bere al giorno e perché è così importante
- Differenze tra apparato urinario maschile e femminile
- Tumore maligno della prostata (carcinoma prostatico): cause, sintomi e terapie
- Le tue feci dicono se sei in salute: con la Scala di Bristol impara ad interpretarle
- PSA totale e free alto: capire i risultati dell’esame e rischio di tumore alla prostata
- Esplorazione rettale digitale della prostata: fa male? A che serve?
- Ipertrofia o iperplasia prostatica benigna: cause, sintomi e cure
- Prostata ingrossata ed infiammata: ecco cosa fare per mantenerla in salute
- Uroflussometria: indicazioni, preparazione, come si esegue
- Prostatite batterica ed abatterica: cause e cure dell’infiammazione della prostata
- Prostata: anatomia, dimensioni, posizione e funzioni in sintesi
- Quanto peso perdiamo ogni volta che andiamo in bagno?
- Stitichezza acuta e cronica: tipi, cause, trattamenti medici e rimedi
- Scura o chiara, liquida o schiumosa: la tua urina rivela la tua salute
- Feci dalla bocca: il vomito fecaloide
- Bruciore e stimoli frequenti di urinare: cistite, sintomi e cure
- Perché la cistite è più frequente nelle donne che negli uomini?
- Differenza tra esame delle urine ed urinocoltura
- Rene: anatomia, funzioni e patologie in sintesi
- Differenza tra surrene e rene
- Differenza tra nefrologo ed urologo: patologie e competenze specifiche e comuni
- Glomerulo renale: schema, funzione e flusso ematico renale
- Com’è fatto un rene? [SCHEMA]
- Azotemia (Urea) alta o bassa: valori, cause, sintomi e cosa fare
- Azotemia alta e reni: cibi da evitare per abbassarla
- Clearance della creatinina: alta o bassa, valori, calcolo e sintomi
- Creatinina alta o bassa: cure e terapie per correggere i valori
- Cos’è il perineo maschile e femminile, dove si trova ed a cosa serve? Perché è così importante per la donna, specie in gravidanza?
- Papilloma vescicale: virus, sintomi, vaccino e cure
- Virus del papilloma (HPV): tipi più pericolosi ed a basso rischio
- Come si contrae il Virus del papilloma (HPV)?
- Infezione da Virus del papilloma, gravidanza e problemi al feto
- Trattamento del Virus del papilloma (HPV)
- Differenza tra uretra e uretere
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
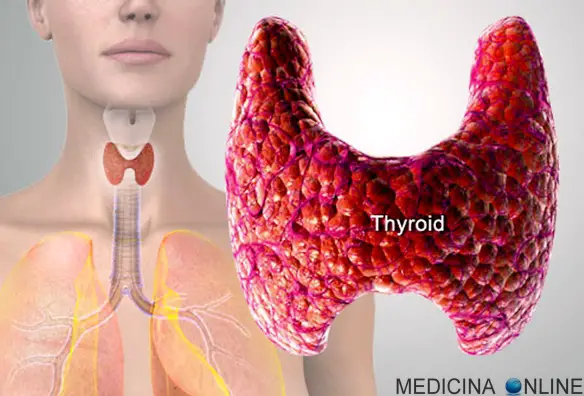 Il carcinoma della tiroide viene considerato una neoplasia rara in quanto costituisce il 2% di tutti i tumori. Si può manifestare a tutte le età, con massima incidenza tra i 25 e i 60 anni e con una maggiore prevalenza nel sesso femminile. Tali neoplasie sono invece molto rare nei bambini. La sopravvivenza è molto elevata, superando il 90% a 5 anni nelle forme differenziate.
Il carcinoma della tiroide viene considerato una neoplasia rara in quanto costituisce il 2% di tutti i tumori. Si può manifestare a tutte le età, con massima incidenza tra i 25 e i 60 anni e con una maggiore prevalenza nel sesso femminile. Tali neoplasie sono invece molto rare nei bambini. La sopravvivenza è molto elevata, superando il 90% a 5 anni nelle forme differenziate. Una terapia si definisce “palliativa” (in inglese “palliative care“) quando non va direttamente a curare la causa della patologia, bensì si occupa di alleviare i sintomi che essa procura al paziente, dando la possibilità a quest’ultimo di godere della migliore qualità di vita possibile, specie in caso di malattia terminale, cioè una malattia che non può essere curata e che condurrà il paziente a morte in un tempo variabile.
Una terapia si definisce “palliativa” (in inglese “palliative care“) quando non va direttamente a curare la causa della patologia, bensì si occupa di alleviare i sintomi che essa procura al paziente, dando la possibilità a quest’ultimo di godere della migliore qualità di vita possibile, specie in caso di malattia terminale, cioè una malattia che non può essere curata e che condurrà il paziente a morte in un tempo variabile. La schizofrenia è una psicosi cronica caratterizzata dalla persistenza di sintomi di alterazione del pensiero, del comportamento e dell’affettività, da un decorso superiore ai sei mesi, con forte disadattamento della persona ovvero una gravità tale da limitare le normali attività di vita della persona.
La schizofrenia è una psicosi cronica caratterizzata dalla persistenza di sintomi di alterazione del pensiero, del comportamento e dell’affettività, da un decorso superiore ai sei mesi, con forte disadattamento della persona ovvero una gravità tale da limitare le normali attività di vita della persona. Il diabete mellito abbreviato DM è una forma di diabete ovvero un gruppo di disturbi metabolici accomunati dal fatto di presentare una persistente instabilità del livello glicemico del sangue, passando da condizioni di iperglicemia, più frequente, a condizioni di ipoglicemia. Sebbene il termine diabete si riferisca nella pratica comune alla sola condizione di diabete mellito come se fossero sinonimi, in realtà esiste un’altra forma di diabete detta diabete insipido, diversa dal diabete mellito. Tali malattie sono accomunate dal solo fatto di presentare abbondanti quantità di urine, non presentando infatti cause, né altri sintomi, comuni. Esistono principalmente due tipi di diabete mellito, il tipo 1 (una volta chiamato insulino-dipendente) ed il tipo 2 (insulino-resistente o insulino-non dipendente).
Il diabete mellito abbreviato DM è una forma di diabete ovvero un gruppo di disturbi metabolici accomunati dal fatto di presentare una persistente instabilità del livello glicemico del sangue, passando da condizioni di iperglicemia, più frequente, a condizioni di ipoglicemia. Sebbene il termine diabete si riferisca nella pratica comune alla sola condizione di diabete mellito come se fossero sinonimi, in realtà esiste un’altra forma di diabete detta diabete insipido, diversa dal diabete mellito. Tali malattie sono accomunate dal solo fatto di presentare abbondanti quantità di urine, non presentando infatti cause, né altri sintomi, comuni. Esistono principalmente due tipi di diabete mellito, il tipo 1 (una volta chiamato insulino-dipendente) ed il tipo 2 (insulino-resistente o insulino-non dipendente). Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard.
Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard.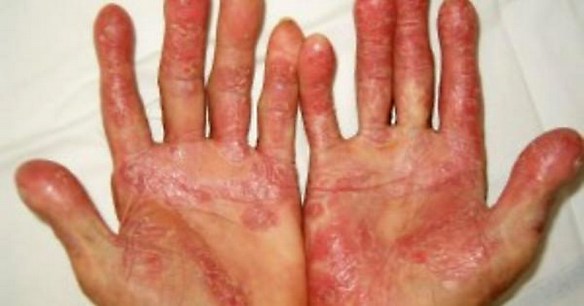 L’artrite psoriasica (AP) è una malattia infiammatoria articolare cronica che si associa una malattia cutanea chiamata psoriasi. Questa malattia è classificata tra le spondiloartriti sieronegative che sono un gruppo di malattie comprendenti anche la spondilite anchilosante, le artriti legate a malattie infiammatorie intestinali (Crohn e colite ulcerosa), le forme indifferenziate (che non rientrano cioè nelle precedenti ).
L’artrite psoriasica (AP) è una malattia infiammatoria articolare cronica che si associa una malattia cutanea chiamata psoriasi. Questa malattia è classificata tra le spondiloartriti sieronegative che sono un gruppo di malattie comprendenti anche la spondilite anchilosante, le artriti legate a malattie infiammatorie intestinali (Crohn e colite ulcerosa), le forme indifferenziate (che non rientrano cioè nelle precedenti ). L’artrite reumatoide (AR) è una poliartrite infiammatoria cronica, anchilosante e progressiva a patogenesi autoimmunitaria e di eziologia sconosciuta, principalmente a carico delle articolazioni sinoviali. Può provocare deformazione e dolore che possono portare fino alla perdita della funzionalità articolare.
L’artrite reumatoide (AR) è una poliartrite infiammatoria cronica, anchilosante e progressiva a patogenesi autoimmunitaria e di eziologia sconosciuta, principalmente a carico delle articolazioni sinoviali. Può provocare deformazione e dolore che possono portare fino alla perdita della funzionalità articolare.