
 Ha solo tre anni, ma bicipiti e addominali da far invidia ai più dotati bodybuilder. Per questo Dash, bambino australiano originario di Canberra, è diventato una vera e propria star dei social network, seguito su Instagram da più di 400mila persone in giro per il mondo impressionate dalle sue incredibili doti fisiche. Il tutto grazie alla mamma Ursula, che all’edizione locale del Daily Mail ha raccontato la trasformazione del figlio in vero e proprio guru della ginnastica nonostante la giovane età.
Ha solo tre anni, ma bicipiti e addominali da far invidia ai più dotati bodybuilder. Per questo Dash, bambino australiano originario di Canberra, è diventato una vera e propria star dei social network, seguito su Instagram da più di 400mila persone in giro per il mondo impressionate dalle sue incredibili doti fisiche. Il tutto grazie alla mamma Ursula, che all’edizione locale del Daily Mail ha raccontato la trasformazione del figlio in vero e proprio guru della ginnastica nonostante la giovane età.
Le foto che circolano online immortalano Dash mentre salta, si arrampica sugli attrezzi in giro per la casa e rinforza i suoi muscoli, che non sono per niente conformi alla sua età. “Anche a me piace fare attività fisica – sottolinea la madre – ma non credete che sia una fanatica dello sport, e neanche mio figlio lo è. Passiamo molto tempo all’aria aperta insieme e ci teniamo in forma”.
Tuttavia, Dash non è sempre stato il bambino in salute che è ora. È nato con due settimane di anticipo e più piccolo rispetto alla norma. Ci ha messo un po’ a mettere peso, ma da quando ha cominciato a camminare, a circa dieci mesi e mezzo, non si è mai fermato, saltando su e giù per i tavoli della cucina e risalendo sulle pareti. “La sua incredibile forza fisica – continua Ursula – viene dall’essere costantemente attivo. Spero che un giorno possa diventare uno sportivo di professione e magari partecipare alle Olimpiadi come ginnasta, ma la cosa importante per ora è che sia in salute e felice”.
Leggi anche:
- Ecco come funzionano i controlli antidoping: il caso Schwazer
- Doping genetico: modifica il DNA col gene dello sprint, della potenza muscolare, della resistenza…
- Morto il bodybuilder Rich Piana a causa degli anabolizzanti
- Ronnie Coleman sulla morte di McCarver: soffocamento? C’è dell’altro
- Ronnie Coleman e la fine della sua carriera di bodybuilder
- Doping genetico: cos’è ed a quale rivoluzione porterà in futuro?
- Nel 2004 era il bambino più muscoloso al mondo, ecco com’è diventato ora
- Il bambino più forte del mondo: bodybuilder a soli 9 anni [VIDEO]
- Italia: secondo paese al mondo per casi di doping
- Brandon Blake, bambino culturista a 9 anni: 15 ore di allenamento
- Doping sempre più diffuso tra i giovani, anche sotto i 12 anni
- Doping a partire dai 7 anni di età: un problema in rapida espansione
- Il doping nei bambini e negli adolescenti: il ruolo dei genitori
- Doping nei bambini: perché è così diffuso e quali farmaci vengono usati
- Le 20 giustificazioni più assurde per i casi di doping nello sport
- Mandibola GH, ormone della crescita e doping nello sport)
- Come riconoscere un atleta “natural” da un dopato in palestra
- Record di doping per un culturista italiano: 14 farmaci proibiti
- Muore a 20 anni per il fisico perfetto: lo uccidono gli steroidi
- Sesso di gruppo ed ecstasy: muore giovane bodybuilder
- Giovane ragazza bodybuilder muore per aver mangiato troppe proteine
- Bodybuilder fa un salto, cade male e muore davanti ai fan [VIDEO]
- Mike Matarazzo: la lettera del bodybuilder morto a causa del doping
- Usa un pene finto per fare pipì pulita e passare il test antidoping
- Lutto nel mondo del culturismo, muore a 33 anni noto bodybuilder salentino
- Bodybuilder perde un braccio per essersi iniettato olio di cocco nei muscoli
- Daniele Seccarecci: morto il bodybuilder dal braccio più grande al mondo
- “Mangiavo solo pizza ed energy drink”: ex bodybuilder muore di tumore
- Un bodybuilder muore e un altro è grave: inchiesta a Foggia
- Morto il bodybuilder di San Donà Enrico Vignotto
- Lutto nel body building: Dallas McCarver è morto a 26 anni
- Hulk Hogan, wrestling e steroidi: “Grazie a Dio sono ancora vivo”
- Bodybuilder 19enne muore per la rottura dell’aorta: usava steroidi anabolizzanti e testosterone
- Steroidi anabolizzanti, sesso e calo della libido: ho smesso ma mi eccito poco
- Andreas Munzer: i dosaggi degli steroidi del bodybuilder morto per emorragia
- “Sono invincibile”: morto a 20 anni ucciso dagli steroidi
- Morto il bodybulder Zyzz: ecco la causa della morte
- Differenza tra integratori alimentari e sostanze dopanti con esempi
- Gli integratori alimentari servono davvero o sono solo moda ed operazioni commerciali?
- Otto brutte conseguenze dell’abuso di steroidi
- Ernestine Shepherd bodybuilder ad 80 anni: “L’età è solo una cifra”
- Sam Bryant: ha 70 anni il bodybuilder più anziano del mondo
- Calciatore segna ed esultando si rompe il crociato
- Ormone della crescita (GH) a che serve e da cosa è prodotto?
- Ormone della crescita (GH): body building e doping in palestra
- Ormone della crescita (GH): effetti avversi nel body building e nello sport
- Ginecomastia: quando è l’uomo ad avere il seno
- Dinitrofenolo (DNP): il ritorno della pillola dietetica mortale
- Synthol per gonfiare i muscoli: cos’è e quali pericoli nasconde
- Synthol nei bicipiti: bodybuilder a rischio amputazione delle braccia
- Differenza tra proteine animali e vegetali: quali sono le migliori?
- Differenza tra proteine ed aminoacidi
- Meglio aminoacidi o proteine?
- Differenza tra aminoacidi essenziali e non essenziali
- Differenza tra aminoacidi essenziali e ramificati
- Quando assumere aminoacidi e ramificati per migliorare l’allenamento
- Io dico NO al doping in palestra, sempre e comunque
- Differenza tra ipertrofia muscolare sarcolplasmatica e miofibrillare
- Ipertrofia muscolare: cosa significa e come si raggiunge
- Differenza tra ipertrofia ed iperplasia con esempi
- Differenza tra iperplasia e neoplasia
- Differenza tra atrofia, distrofia ed aplasia con esempi
- Differenza tra omega 3, omega 6 ed omega 9: quale integratore scegliere?
- Differenza tra grassi animali e vegetali
- Differenza tra grassi ed oli con esempi
- Differenza grassi saturi, monoinsaturi e polinsaturi
- Integratore di acido alfa lipoico: a che serve e qual è il dosaggio?
- Differenze tra acido alfa lipoico ed acido linoleico coniugato
- Differenza tra creatina monoidrato ed alcalina: quale preferire?
- Differenza tra creatina e carnitina: quale delle due preferire?
- Differenze proteine whey concentrate, isolate e idrolizzate: quale scegliere?
- Proteine whey: quando, quante e perché assumerle?
- Creatina e sport: quando, quanta e perché assumerla?
- Creatina e caffè o coca cola: posso assumerli insieme?
- I 6 principi base dell’allenamento in palestra
- Allenamento ad alta intensità e cedimento muscolare per spingerti oltre ogni limite
- Il recupero tra le serie non va fatto a caso: ogni allenamento ha il suo recupero ideale
- Allenamento in palestra: gli errori che fanno spesso gli uomini
- Cosa e come mangiare prima dell’allenamento?
- Phil Heath: allenamento, dieta ed integratori del grande bodybuilder
- Core stability ed allenamento in palestra
- Allenamento con le ripetute (interval training): cos’è ed a che serve?
- Nuoto e triathlon: quanto conta e come allenarsi
- Cause di fatica muscolare: acidificazione del muscolo e deplezione del glicogeno muscolare
- L’altezza media italiana nel 2017 di maschi e femmine
- L’altezza media italiana 2017 da 1 a 19 anni di maschi e femmine
- L’altezza media mondiale nel 2017 di maschi e femmine [TABELLA]
- Altezza: quando si può parlare di nanismo o gigantismo
- Quanto è alto l’uomo più alto del mondo?
- Quanto è alto l’uomo più basso del mondo?
- Nanismo: sintomi, cura, cause, terapia, diagnosi e prevenzione
- Charlotte, la bambina più piccola del mondo
- Allenamento: in palestra non fare MAI questi errori
- Intensità, volume, cedimento, cadenza e pause tra gli esercizi: le 5 variabili dell’allenamento coi pesi
- Costruire un fisico muscoloso: i 14 punti chiave
- Pre-esaurimento muscolare: cos’è, a che serve e schemi pratici
- Come aumentare la massa dei pettorali senza panca piana
- Recupero breve tra le serie per aumentare la definizione muscolare
- Allungamento muscolare forzato per aumentare la massa muscolare: scheda di allenamento
- Differenza tra contrazione eccentrica e concentrica con esempi
- Differenza tra contrazione isometrica (statica) e isotonica (dinamica) con esempi
- Schemi di allenamento, riposo alimentazione, integrazione: come raggiungere la super compensazione
- Gli integratori alimentari: fanno bene o fanno male?
- Gli integratori migliori per aumentare massa e definizione muscolare
- Integratori alimentari nello sport: guida facile per principianti
- I 7 integratori alimentari necessari per chi segue una dieta vegana
- Bicarbonato di sodio: come influenza la prestazione sportiva?
- Arginina per aumentare il pump muscolare
- Quali sono gli integratori alimentari migliori per il crossfit?
- I 4 integratori più sottovalutati ma che invece sono molto utili nello sport
- I 5 integratori economici che aumentano le vostre prestazioni
- Creatina monoidrato ed acido linoleico coniugato (CLA) per migliorare le tue prestazioni
- Integratori soppressori dell’appetito per perdere peso
- Integratori: quando assumerli per potenziarne gli effetti?
- Carnosina e β-alanina per incrementare la performance sportiva
- Semi di lino e olio di lino: benefici reali e teorici
- Non sono grasso: ho la costituzione robusta e le ossa grosse
- Differenza tra ginecomastia vera, falsa, acquisita, congenita e puberale
- Quando prendere le proteine per potenziarne l’effetto?
- Differenza tra fibre muscolari bianche e rosse
- Potenziale genetico: come calcolare la massima crescita muscolare possibile
- Scala dello sforzo percepito (RPE) e percentuali di carico nell’allenamento con i pesi
- Il segreto per progredire nel tuo allenamento? La periodizzazione
- Whey o aminoacidi essenziali: quale scegliere per aumentare la massa muscolare?
- Frequenza dei pasti e matabolismo: meglio pasti piccoli e frequenti o pochi ed abbondanti
- Pasto libero: “sgarrare” nella dieta è davvero così deleterio?
- Intolleranza al glutine e sport: gestione dell’atleta celiaco
- I carboidrati fanno davvero ingrassare?
- Quante proteine assumere dopo l’allenamento con i pesi (post-workout)?
- Ricomposizione corporea e rapporto tra massa grassa e magra
- I grassi si bruciano dopo 20 minuti di attività: vero o falso?
- La palestra e lo sport possono cambiare i lineamenti del volto?
- Differenza tra proteine caseine e whey
- Differenza tra pesi, kettlebell e bilanciere: punti deboli e di forza
- Differenza tra pesi e corpo libero: vantaggi e svantaggi
- È meglio mangiare o no prima di un allenamento?
- Esercizi a corpo libero: come diventare “grossi” senza i pesi
- Differenza tra pesi ed elastici: vantaggi e svantaggi delle due tecniche
- Le 5 cose che NON devi MAI fare dopo aver finito l’allenamento
- Palestra: ad obiettivi diversi corrisponde una diversa alimentazione
- Corsa, frequenza cardiaca, formula di Tanaka e cardiofrequenzimetro
- Le 10 ragioni per cui non devi MAI dimenticarti di riscaldarti prima dell’allenamento
- Non solo ipertensione: ecco gli effetti del troppo sale nella dieta
- Allenamento con la palla medica: perché è importante e quali sono i vantaggi
- Allenarsi troppo non fa dimagrire
- Come e quanto correre per dimagrire?
Lo staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o seguici su Twitter, su Instagram o su Pinterest, grazie!
 Con il termine “epilessia” ci si riferisce ad un insieme di disturbi cerebrali che vanno da forme molto gravi, pericolose per la vita e invalidanti, a forme molto più benigne. In un soggetto affetto da questo disturbo la funzione neuronale (del sistema nervoso) viene disturbata e alterata, andando a causare anomalie in
Con il termine “epilessia” ci si riferisce ad un insieme di disturbi cerebrali che vanno da forme molto gravi, pericolose per la vita e invalidanti, a forme molto più benigne. In un soggetto affetto da questo disturbo la funzione neuronale (del sistema nervoso) viene disturbata e alterata, andando a causare anomalie in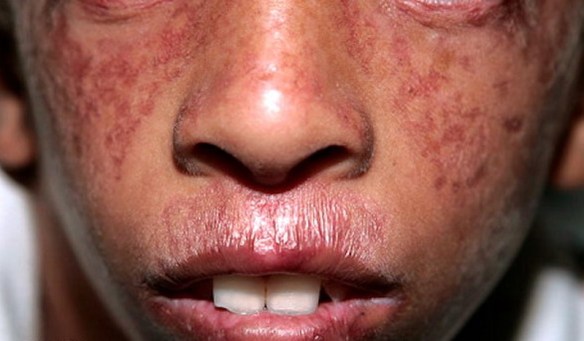 La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie.
La sindrome di Bloom è una malattia autosomica recesssiva caratterizzata da eritema teleangiectasico del volto, fotosensibilita, nanismo ed altre anomalie. Spesso i neo-genitori si spaventano per qualche particolare delle feci del proprio bimbo che sembrano anomale, ma sono invece del tutto normali. Vediamo allora che caratteristiche hanno le feci del neonato, come cambiano nel tempo e quando è il caso di preoccuparsi.
Spesso i neo-genitori si spaventano per qualche particolare delle feci del proprio bimbo che sembrano anomale, ma sono invece del tutto normali. Vediamo allora che caratteristiche hanno le feci del neonato, come cambiano nel tempo e quando è il caso di preoccuparsi.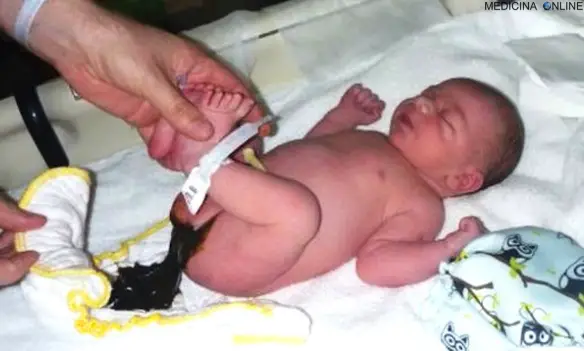
 La sindrome di Down è un’anomalia cromosomica che si manifesta attraverso diversi sintomi congeniti: le persone affette sono caratterizzate da una copia in eccesso del cromosoma 21 e possono manifestare sintomi fisici e disabilità intellettive. Tutti i casi diagnosticati presentano un ritardo cognitivo, ma il grado di disabilità è molto variabile tra gli individui affetti, con la maggior parte che rientra nella gamma di “poco” o “moderatamente disabili”. Purtroppo i soggetti Down spesso sono affetti da altri problemi di salute, per esempio malformazioni cardiache, difetti dell’udito, intestinali, tiroidei, scheletrici e agli occhi. La gravità di questi problemi varia considerevolmente a seconda dell’individuo. La sindrome è la più comune anomalia cromosomica nell’uomo e il rischio di concepire un bimbo Down aumenta proporzionalmente all’età della madre. La diagnosi può avvenire al momento della nascita o anche prima, attraverso lo screening prenatale. Questa sindrome non può essere curata, anche se un approccio integrato impostato fin dai primi mesi di vita permette di favorire un buon sviluppo delle capacità di base, permettendo di condurre una vita serena e produttiva. Negli ultimi decenni l’aspettativa di vita è aumentata significativamente, passando dai 25 anni del 1983 ai 60 e più attuali.
La sindrome di Down è un’anomalia cromosomica che si manifesta attraverso diversi sintomi congeniti: le persone affette sono caratterizzate da una copia in eccesso del cromosoma 21 e possono manifestare sintomi fisici e disabilità intellettive. Tutti i casi diagnosticati presentano un ritardo cognitivo, ma il grado di disabilità è molto variabile tra gli individui affetti, con la maggior parte che rientra nella gamma di “poco” o “moderatamente disabili”. Purtroppo i soggetti Down spesso sono affetti da altri problemi di salute, per esempio malformazioni cardiache, difetti dell’udito, intestinali, tiroidei, scheletrici e agli occhi. La gravità di questi problemi varia considerevolmente a seconda dell’individuo. La sindrome è la più comune anomalia cromosomica nell’uomo e il rischio di concepire un bimbo Down aumenta proporzionalmente all’età della madre. La diagnosi può avvenire al momento della nascita o anche prima, attraverso lo screening prenatale. Questa sindrome non può essere curata, anche se un approccio integrato impostato fin dai primi mesi di vita permette di favorire un buon sviluppo delle capacità di base, permettendo di condurre una vita serena e produttiva. Negli ultimi decenni l’aspettativa di vita è aumentata significativamente, passando dai 25 anni del 1983 ai 60 e più attuali. Togliamoci subito ogni dubbio. Il Parmigiano Reggiano è un toccasana per la dieta quotidiana. Non solo per quella di adulti e anziani, ma pure per quella di bambini e adolescenti, che hanno bisogno del giusto apporto di calcio per crescere sani e forti.
Togliamoci subito ogni dubbio. Il Parmigiano Reggiano è un toccasana per la dieta quotidiana. Non solo per quella di adulti e anziani, ma pure per quella di bambini e adolescenti, che hanno bisogno del giusto apporto di calcio per crescere sani e forti. Latte materno e latte artificiale: quali differenze?
Latte materno e latte artificiale: quali differenze?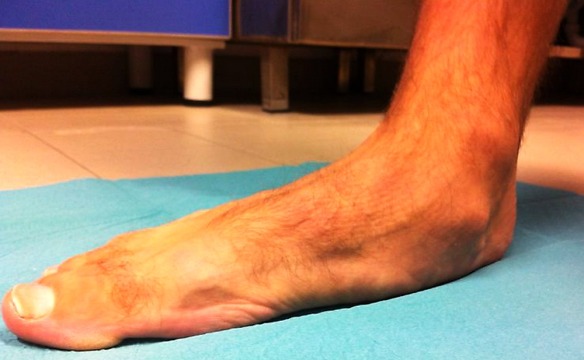 Con “piede piatto” viene indicata una particolare conformazione del piede caratterizzata dall’appiattimento della volta plantare (la parte della superficie plantare del piede che, in situazioni fisiologiche, non tocca il terreno quando si è in posizione eretta) e dalla valgo-pronazione del calcagno. E’ quindi un paramorfismo in cui risultano alterati i rapporti anatomici del piede.
Con “piede piatto” viene indicata una particolare conformazione del piede caratterizzata dall’appiattimento della volta plantare (la parte della superficie plantare del piede che, in situazioni fisiologiche, non tocca il terreno quando si è in posizione eretta) e dalla valgo-pronazione del calcagno. E’ quindi un paramorfismo in cui risultano alterati i rapporti anatomici del piede.