
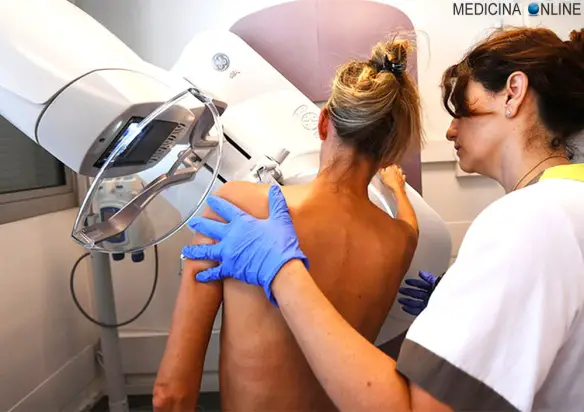
 Le donne dispongono di strumenti molto efficaci per la diagnosi precoce del tumore del seno, tra essi sono di estrema importanza quelli appartenenti al campo della diagnostica per immagini, primo tra tutti la mammografia, affiancata da altri quali ecografia, risonanza magnetica e PET. La prevenzione è fondamentale perché individuare un tumore ancora molto piccolo aumenta notevolmente la possibilità di curarlo in modo definitivo, ma è importante scegliere lo strumento più adatto a noi, fermo restando che una visita senologica periodica è sempre indicata in tutte le donne al di sopra dei 20 anni, specie se hanno casi di tumore alla mammella in famiglia. Ricordiamo che per le donne positive al test genetico per BRCA1 o 2 è indicata un’ecografia semestrale ed una risonanza annuale, anche in giovane età.
Le donne dispongono di strumenti molto efficaci per la diagnosi precoce del tumore del seno, tra essi sono di estrema importanza quelli appartenenti al campo della diagnostica per immagini, primo tra tutti la mammografia, affiancata da altri quali ecografia, risonanza magnetica e PET. La prevenzione è fondamentale perché individuare un tumore ancora molto piccolo aumenta notevolmente la possibilità di curarlo in modo definitivo, ma è importante scegliere lo strumento più adatto a noi, fermo restando che una visita senologica periodica è sempre indicata in tutte le donne al di sopra dei 20 anni, specie se hanno casi di tumore alla mammella in famiglia. Ricordiamo che per le donne positive al test genetico per BRCA1 o 2 è indicata un’ecografia semestrale ed una risonanza annuale, anche in giovane età.
Ecografia
L’ecografia è lo strumento più adatto per la diagnosi quando la paziente ha meno di 35/40 anni e/o ha una mammella di dimensione ridotta (con scarsa componente adiposa), la mammografia è meno raccomandata perché la struttura troppo densa del tessuto mammario in questa fascia di età renderebbe poco chiari i risultati. Ciò è determinato dal fatto che, per motivi legati alla restituzione dell’immagine, l’ecografia permette di trovare lesioni più facilmente quando la mammella è giovane (maggiore componente ghiandolare), mentre la mammografia permette la localizzazione di lesioni quando è maggiore la componente adiposa (pazienti meno giovani). Adipe e noduli hanno una ecogenicità simile, quindi una mammella fortemente adiposa renderebbe infatti più difficile riconoscere il nodulo.
In situazioni particolari, per esempio in caso di scoperta di noduli sospetti, è possibile approfondire l’analisi ecografica con una biopsia (agoaspirato) del nodulo e successiva indagine istologica.
L’ecografia ha molti vantaggi: per prima cosa è indolore, economica e lo strumento ecografico è facilmente reperibile in qualsiasi struttura sanitaria anche piccola. Un grande pregio della ecografia rispetto ad altre metodiche è che permette di visualizzare una struttura in tempo reale con molti vantaggi (si pensi ad esempio alla visualizzazione della reazione del nodulo alla mobilizzazione che – se presente – è indice di benignità), inoltre l’ecografia non utilizza radiazioni ionizzanti come invece avviene con la mammografia, ciò la rende utile nello studio di tessuti in una donna incinta e lo rende un esame ripetibile molte volte, senza alcun rischio per la paziente.
Leggi anche:
- Tumore alla mammella: cause, diagnosi e prevenzione
- La visita senologica: come, quando e perchè farla?
- Riconoscere il cancro al seno: sintomi precoci e tardivi
- Tumore al seno: sintomi e segni visibili
Mammografia
La mammografia è lo strumento più adatto per la diagnosi quando la paziente ha più di 35/40 anni e/o ha una mammella di grandi dimensioni (con adipe maggiormente sviluppato). In questi casi l’ecografia è meno indicata perché – come già prima accennato – una mammella di una donna meno giovane ha solitamente una forte componente adiposa che rende difficile distinguere noduli sospetti in virtù di simile ecogenicità tra adipe e nodulo. Nella mammografia invece adipe e nodulo vengono restituiti con una “luminosità” differente e ciò rende il nodulo più facilmente distinguibile in una mammella con forte componente adiposa.
In caso di dubbio, è comunque certamente utile affiancare una ecografia alla mammografia. E’ importante ricordare che tra i 50 e i 60 anni il rischio di sviluppare un tumore del seno è piuttosto alto e di conseguenza le donne in questa fascia di età devono sottoporsi a controllo mammografico ogni anno. E’ anche importante ricordare che la mammografia, al contrario dell’ecografia, utilizza radiazioni ionizzanti e ciò la rende un esame difficilmente ripetibile, specie se la paziente è incinta.
Anche se la mammografia rimane uno strumento molto efficace per la diagnosi precoce del tumore del seno, oggi sono disponibili anche altre tecniche diagnostiche come la risonanza magnetica (ancora limitata a casi selezionati), la PEM (una tomografia a emissione di positroni – PET – specifica per le mammelle) e un nuovo esame già definito il Pap-test del seno che consiste nell’introduzione di liquido nei dotti galattofori (i canali attraverso i quali passa il latte) e nella successiva raccolta di questo liquido che porta con sé anche alcune cellule. Grazie al microscopio è poi possibile individuare quali tra le cellule fuoriuscite ha caratteristiche pretumorali permettendo una diagnosi molto precoce del tumore del seno.
Conclusioni
La mammografia è indicata in particolare dopo i 35/40 anni (o comunque nelle donne con adipe mammario molto sviluppato). Il seno, infatti, cambia con l’età: aumenta il tessuto adiposo – che appare scuro alla mammografia – mentre diminuisce il volume della ghiandola, che appare chiara. Su fondo scuro, quindi, ogni formazione sospetta diventa immediatamente visibile. Nelle donne più giovani, invece, in genere si verifica la situazione contraria: la massa della ghiandola (chiara) prevale sull’adipe (scuro), perciò è più difficile notare eventuali formazioni, specialmente se molto piccole. L’ecografia – mostrando invece gradazioni di grigio invertite rispetto alla mammografia – risulta l’opzione migliore nelle donne più giovani (prima dei 35/40 anni) o comunque in quelle con massa ghiandolare che prevale su quella adiposa.
Leggi anche:
- Biopsia del linfonodo sentinella: a che serve, perché è importante
- La mammografia: un esame rapido che può salvarti la vita
- L’ecografia mammaria: un esame innocuo ed indolore che ti può salvare vita
- Cosa sono le metastasi? Tutti i tumori danno metastasi?
- Cancro al seno: sintomi precoci, diagnosi, terapia e prevenzione
- Tumore al seno: stadiazione, prognosi e sopravvivenza
- Cancro al seno: metastasi e sintomi avanzati del tumore
- Noduli al seno: quando preoccuparsi ed andare dal medico?
- Come riconoscere un nodulo maligno del seno da uno benigno?
- Tumore al seno: sintomi e dolore al braccio
- Tumore al seno C1 C2 C3 C4 C5: cosa significa il referto?
- Tumore al seno età: a quanti anni si può verificare?
- Classificazione e stadiazione delle fasi del tumore alla mammella
- Capezzolo retratto (introflesso): cause, cancro al seno e trattamenti
- Capezzolo dolorante e sensibile in uomo, donna, gravidanza e menopausa
- Malattia di Paget del capezzolo: sintomi precoci, cause e cure
- Mammella: anatomia e funzioni del seno e delle ghiandole mammarie
- Divisione in quadranti della mammella (Q1 Q2 Q3 Q4)
- Quadranti mammari, tumore al seno, quadrantectomia e mastectomia radicale
- Breast Unit salvavita: -18% di mortalità in caso di cancro al seno
- Capezzolo femminile: forma, dimensioni, funzioni e simmetria
- Differenza dei capezzoli e del seno in gravidanza
- A cosa serve e come si fa l’autopalpazione del seno?
- Autopalpazione della mammella: come farla nel modo giusto [VIDEO]
- Perché ho il seno piccolo? Quali sono i fattori che influenzano la grandezza del mio seno?
- Ginecomastia: quando è l’uomo ad avere il seno
- Differenza tra ginecomastia vera, falsa, acquisita, congenita e puberale
- L’uomo può allattare al seno?
- Storia di un seno: dall’embrione alla menopausa
- Cos’è la pubertà, a che età inizia e come si manifesta?
- Il reggiseno provoca il cancro al seno? Finalmente abbiamo una risposta
- Perché agli uomini piace così tanto il seno delle donne?
- Ormoni estrogeni: cosa sono e quali funzioni svolgono?
- Differenza dolore al seno da gravidanza e da ciclo mestruale
- Differenze tra neonati allattati con latte materno ed artificiale
- Quando una mammella non si sviluppa: la Sindrome di Poland
- Politelia: quando i capezzoli sono troppi, cause e terapie
- Mastodinia: quando il seno è gonfio e dolorante
- Rassodare il seno senza chirurgia con la Radiofrequenza Monopolare
- Aumentare il seno senza chirurgia con i cibi ricchi di fitoestrogeni
- Vuoi avere un seno più bello? Comincia con la postura e la ginnastica giusta
- Dimagrire senza perdere seno
- Prendere il sole al seno fa male? Come abbronzarlo in sicurezza
- Grandezza del seno: le donne del nord battono quelle del sud
- La donna con il seno più grande del mondo [VIDEO]
- I deodoranti che contengono alluminio causano il cancro al seno?
- Esercizi e consigli per rassodare e tonificare il seno
- Il reggiseno che può far crescere il tuo seno
- L’incidenza del tumore al seno in crescita tra le quarant’enni: l’importanza della mammografia
- Linfonodo sentinella: cos’è e perché è importante in caso di cancro
- Rossore ed irritazione della pelle sotto e tra il seno: cause e rimedi
- Fitoestrogeni: rimedi naturali in menopausa
- Ingrandire il seno con l’ipnosi
- Filler ad alta densita’: Macrolane™ per l’aumento del seno senza bisturi
- I fitoestrogeni aumentano il rischio di cancro al seno?
Dott. Emilio Alessio Loiacono
Medico Chirurgo
Direttore dello Staff di Medicina OnLine
Se ti è piaciuto questo articolo e vuoi essere aggiornato sui nostri nuovi post, metti like alla nostra pagina Facebook o unisciti al nostro gruppo Facebook o ancora seguici su Twitter, su Instagram, su Mastodon, su YouTube, su LinkedIn, su Tumblr e su Pinterest, grazie!
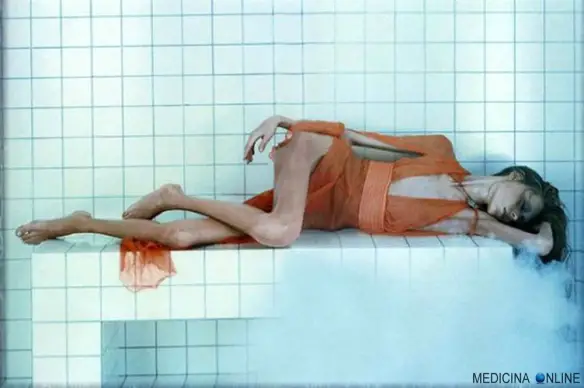 Quando una persona ha una forte perdita di peso rispetto al peso forma, potrebbe in alcuni casi essere portata a chiedersi se è sottopeso o anoressica. Nel caso della donna che vedete nella foto in alto, è difficile essere nel dubbio: quando il dimagrimento è davvero eccessivo, è anoressia nervosa. Ma se il dimagrimento è più sfumato, come capire se si è “solo” sottopeso o se siamo anoressici? Tutte le persone in sottopeso sono anoressiche? Facciamo chiarezza.
Quando una persona ha una forte perdita di peso rispetto al peso forma, potrebbe in alcuni casi essere portata a chiedersi se è sottopeso o anoressica. Nel caso della donna che vedete nella foto in alto, è difficile essere nel dubbio: quando il dimagrimento è davvero eccessivo, è anoressia nervosa. Ma se il dimagrimento è più sfumato, come capire se si è “solo” sottopeso o se siamo anoressici? Tutte le persone in sottopeso sono anoressiche? Facciamo chiarezza. La schizofrenia è una psicosi cronica caratterizzata dalla persistenza di sintomi di alterazione del pensiero, del comportamento e dell’affettività, da un decorso superiore ai sei mesi, con forte disadattamento della persona ovvero una gravità tale da limitare le normali attività di vita della persona.
La schizofrenia è una psicosi cronica caratterizzata dalla persistenza di sintomi di alterazione del pensiero, del comportamento e dell’affettività, da un decorso superiore ai sei mesi, con forte disadattamento della persona ovvero una gravità tale da limitare le normali attività di vita della persona. Il carcinoma della tiroide viene considerato una neoplasia rara in quanto costituisce il 2% di tutti i tumori. Si può manifestare a tutte le età, con massima incidenza tra i 25 e i 60 anni e con una maggiore prevalenza nel sesso femminile. Tali neoplasie sono invece molto rare nei bambini. La sopravvivenza è molto elevata, superando il 90% a 5 anni nelle forme differenziate.
Il carcinoma della tiroide viene considerato una neoplasia rara in quanto costituisce il 2% di tutti i tumori. Si può manifestare a tutte le età, con massima incidenza tra i 25 e i 60 anni e con una maggiore prevalenza nel sesso femminile. Tali neoplasie sono invece molto rare nei bambini. La sopravvivenza è molto elevata, superando il 90% a 5 anni nelle forme differenziate. La defecografia è un esame radiologico utilizzato per studiare le disfunzioni del pavimento pelvico che causano problemi nella defecazione. Viene eseguito utilizzando un mezzo di contrasto baritato diluito, iniettato direttamente nell’ampolla rettale. In alcuni casi , se richiesto, vengono opacizzate anche le anse del piccolo intestino con mezzo di contrasto baritato per bocca, la vescica mediante mezzo di contrasto iodato posizionando catetere vescicale o anche la vagina (nelle donne) per studiare i rapporti reciproci degli organi opacizzati durante la defecazione.
La defecografia è un esame radiologico utilizzato per studiare le disfunzioni del pavimento pelvico che causano problemi nella defecazione. Viene eseguito utilizzando un mezzo di contrasto baritato diluito, iniettato direttamente nell’ampolla rettale. In alcuni casi , se richiesto, vengono opacizzate anche le anse del piccolo intestino con mezzo di contrasto baritato per bocca, la vescica mediante mezzo di contrasto iodato posizionando catetere vescicale o anche la vagina (nelle donne) per studiare i rapporti reciproci degli organi opacizzati durante la defecazione. Il diabete mellito abbreviato DM è una forma di diabete ovvero un gruppo di disturbi metabolici accomunati dal fatto di presentare una persistente instabilità del livello glicemico del sangue, passando da condizioni di iperglicemia, più frequente, a condizioni di ipoglicemia. Sebbene il termine diabete si riferisca nella pratica comune alla sola condizione di diabete mellito come se fossero sinonimi, in realtà esiste un’altra forma di diabete detta diabete insipido, diversa dal diabete mellito. Tali malattie sono accomunate dal solo fatto di presentare abbondanti quantità di urine, non presentando infatti cause, né altri sintomi, comuni. Esistono principalmente due tipi di diabete mellito, il tipo 1 (una volta chiamato insulino-dipendente) ed il tipo 2 (insulino-resistente o insulino-non dipendente).
Il diabete mellito abbreviato DM è una forma di diabete ovvero un gruppo di disturbi metabolici accomunati dal fatto di presentare una persistente instabilità del livello glicemico del sangue, passando da condizioni di iperglicemia, più frequente, a condizioni di ipoglicemia. Sebbene il termine diabete si riferisca nella pratica comune alla sola condizione di diabete mellito come se fossero sinonimi, in realtà esiste un’altra forma di diabete detta diabete insipido, diversa dal diabete mellito. Tali malattie sono accomunate dal solo fatto di presentare abbondanti quantità di urine, non presentando infatti cause, né altri sintomi, comuni. Esistono principalmente due tipi di diabete mellito, il tipo 1 (una volta chiamato insulino-dipendente) ed il tipo 2 (insulino-resistente o insulino-non dipendente). Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard.
Con nanismo si intende una situazione patologica caratterizzata dal mancato raggiungimento del livello staturale della media della popolazione. Si distinguono nanismi armonici e nanismi disarmonici. Più precisamente, si parla di nanismo quando l’altezza di un individuo risulta inferiore di tre deviazioni standard sulla curva di accrescimento normale stabilita in funzione dell’età e del sesso. Esistono infatti tabelle di accrescimento, elaborate a partire da ampie popolazioni infantili, che forniscono, per ogni età e sesso, un’altezza media normale ed il valore di una deviazione standard. Diagnosi
Diagnosi